
13: Biochemistry, Climate Change and Human Health
- Page ID
- 34468
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Search Fundamentals of Biochemistry
(Learning goals written by Claude, Anthropic)
By the end of this chapter, students should be able to:
One Health, Heat Illness, and Kidney Disease
- Explain the One Health framework and describe the spectrum of heat-related illness (cramps, exhaustion, syncope, heat stroke), connect increasing heat wave frequency and severity to mortality data from major events (2003 Europe: 70,000; 2010 Russia: 55,000; 2022 Europe: 60,000), and explain why cardiovascular strain develops at 34°C in humid versus 41°C in dry conditions.
- Describe the clinical presentation, geographic distribution, and proposed mechanism of CKDu — connecting occupational heat/humidity exposure to renal inflammation and fibrosis in the absence of traditional risk factors — and identify its global expansion from Mesoamerica to Sri Lanka, India, Florida, and California.
Necroptosis, ZBP1, and Heat Stroke Mechanisms
- Describe the necroptosis pathway from receptor activation through RIPK3 autophosphorylation (pT224, pS227) to MLKL membrane translocation, calcium influx, and membrane permeabilization, contrasting this caspase-independent pathway with classical apoptosis and identifying the regulatory phosphorylation sites (pS164, pT165) that divert signaling toward apoptosis.
- Explain how ZBP1 activates RIPK3 through its RHIM domain during heat stress — independently of its Zα nucleic acid-binding domain — citing the experimental evidence (RHIM mutations prevent death; Zα mutations do not; ZBP1-GFP aggregation through RHIM), and distinguish this from viral Z-RNA/Z-DNA sensing.
- Distinguish necroptosis, pyroptosis (gasdermin pore formation, IL-1β/IL-18 release), and PANoptosis, and explain how heat stroke-induced epigenetic changes in monocyte DNA methylation patterns persist for 30+ days and are inherited by daughter cells, providing a mechanism for reduced heat tolerance after prior illness.
PM2.5: Sources, Entry, and Cellular Toxicity
- Define PM10, PM2.5, and PM0.1, describe the chemical composition of PM2.5 particles (black carbon, PAHs, transition metals, sulfate/nitrate/ammonium), and explain how secondary pollutants form — including tropospheric ozone from NOx/hydrocarbons and secondary organic aerosols from α-pinene/limonene terpene photochemical oxidation.
- Describe the three entry routes for PM2.5 (phagocytosis, pinocytosis, clathrin/caveolin endocytosis), explain how nanoscale PM0.1 protein coronas (hemoglobin, albumin, fibrinogen) amplify lung fibroblast proliferation and fibrosis, and explain the chemistry of the 1952 London Fog (SO₂ + NO₂ → sulfate in aqueous droplets, concentrated by evaporation) that killed ~12,000 people in two weeks.
- Explain the NRF2/Keap1/p62 antioxidant axis — how electrophilic PM2.5 metabolites modify Keap1 Cys residues releasing NRF2, how p62 accumulation creates a positive feedback loop, and how PM2.5 lysosomal membrane permeabilization and organelle damage ultimately activate apoptosis, necroptosis, and pyroptosis — and connect long-term PM2.5 exposure to neural damage (increased phospho-tau, malondialdehyde in hippocampus) and to epithelial-mesenchymal transition (EMT) via TGF-β/SMAD, PI3K/Akt, Wnt/β-catenin, and NF-κB signaling as a mechanism for lung cancer.
Introduction
Climate change affects human and biospheric health, which are inextricably linked. Many, including the US CDC, define One Health as "a collaborative, multisectoral, and transdisciplinary approach—working at the local, regional, national, and global levels—to achieve optimal health outcomes recognizing the interconnection between people, animals, plants, and their shared environment."
We all know that pollution from the use of fossil fuels also has severe health consequences independent of effects mediated more directly by climate change. A solution to both is to dramatically decrease the use of fossil fuels and mitigate pollution from their use. People most likely do not understand the extent to which climate change and fossil fuel use are linked to human health. If they did, they might become advocates for climate action. This section will cover how climate change and fossil fuel use affect human health and diseases. This section will focus on heat-related illnesses and pulmonary/cardiovascular diseases. The next chapter will address climate change, emerging diseases, and pandemics.
Fossil fuel use and climate change affect other diseases, including some indirectly. For instance, increases in cancer deaths will occur due to lower availability of health care arising from extreme weather disasters that impact health facilities and people's access to them. An increase in cancer deaths occurred during the COVID pandemic, since people deferred preventive healthcare treatments as well as cancer surgeries during the pandemic. Allergic illness will increase as growing seasons lengthen and species that cause allergic reactions shift to new growth regions.
Heat-Related Illness
Heat illnesses include cramps, exhaustion, syncope (fainting), and heat stroke, the latter of which can quickly become fatal. Heat affects normal physiology and health. About 1% of all cardiovascular deaths are estimated to be linked to extreme temperatures. We know the number of warm days and heat waves has increased with climate change.
Figure \(\PageIndex{1}\) below shows how heat waves have changed in the US in the decades from 960 to 2022 using data from 50 large metropolitan areas. The 2020s are on track to continue the trend of increasing heat waves.

Figure \(\PageIndex{1}\): Climate Change Indicators: Heat Waves. EPA. https://www.epa.gov/climate-indicato...ors-heat-waves. Data source: NOAA (2022)
Many recent historical heat waves have occurred. A heat wave in Europe in 2003 caused 15,000 heat-exposure-related deaths in France and 70,000 throughout Europe. On June 28, 2019, France recorded a temperature of 45.9 °C (115 °F). In 2022, China experienced a two-month heat wave and drought, with a record temperature of 45 °C (113 °F) in Chongqing. The Chicago Heat Wave of 1995, with a maximal temperature of 106 °F, caused by high temperatures and humidity, killed over 500 people. The worst might be the Russian Heat Wave of 2010, when temperatures were 5 °C (9 °F) higher than normal and reached 40 °C (104 °F). Around 55,000 people died.
Figure \(\PageIndex{2}\) shows temperature and excess mortality from the 2022 heat wave in Europe, during which 60,000 people died from heat-related causes.

Figure \(\PageIndex{2}\): Weekly temperature and heat-related mortality numbers in Europe during the summer of 2022. Ballester, J., Quijal-Zamorano, M., Méndez Turrubiates, R.F. et al. Heat-related mortality in Europe during the summer of 2022. Nat Med (2023). https://doi.org/10.1038/s41591-023-02419-z. Creative Commons Attribution 4.0 International License. http://creativecommons.org/licenses/by/4.0/
Panel a shows the weekly baseline (gray line) and observed (black line) temperature (°C) averaged over Europe. Temperature anomalies are the differences between observed and baseline temperatures (gray shading). Baseline temperatures were computed as the mean annual cycle of observed temperatures in the reference period 1991–2020.
Panels b and c show weekly heat-related mortality (weekly deaths) aggregated over Europe for the overall population (black), women (red), and men (blue) (b) and people aged 0–64 (blue), 65–79 (red), and 80+ (black) years (c), together with their 95% CIs (shadings). The numbers for women and men in b do not include the United Kingdom; values for the age groups in c do not include Germany, Ireland, and the United Kingdom.
Figure \(\PageIndex{3}\) below shows the steady increase in deaths as the average summer temperature in Europe has increased. The 2022 outlier is shown as a red dot.

Figure \(\PageIndex{3}\): The summer of 2022 within the context of rising temperatures in Europe. Relationship between summer mean temperature (°C) and summer heat-related mortality (summer deaths) in the analyzed European countries. The straight line shows the linear fitting for the 2015–2022 period. Ballester et al., ibid.
In exercise studies under controlled conditions, the heart rate increases and plateaus after a temperature increase. If the temperature increases further, the heart rate increases with less plateauing, a sign of cardiovascular strain. In humid conditions, cardiovascular strain develops even on slow walking at 34 °C (93.2 °F). Under dry conditions, the strain develops at around 41 °C (106 °F). The strain (as indicated by an increasing heart rate) proceeds for about 20 minutes, resulting in a rise in core temperature.
The annual probably of heat waves with apparent temperatures of 40 °C/104 °F (dangerous with a high incidence of heat cramps, heat exhaustion, and heat strokes) and 55 °C./131 °F (very dangerous with heat stoke very likely) increases as the average global temperatures increase from the pre-Industrial Revolution values by 1.5 - 4 °C (y-axis), as shown in Figure \(\PageIndex{4}\) below.

Figure \(\PageIndex{4}\): Annual probability of occurrence of heat waves with apparent temperature (with contributions from humidity) peaks greater than 40 °C and 55 °C. Russo, S., Sillmann, J. & Sterl, A. Humid heat waves at different warming levels. Sci Rep 7, 7477 (2017). https://doi.org/10.1038/s41598-017-07536-7. Creative Commons Attribution 4.0 International License. http://creativecommons.org/licenses/by/4.0/.
Panels (a–c) show the probability of occurrence of heat waves with AT peak ≥ 40 (AT40C) calculated at each grid point for all model years with global mean temperature anomaly relative to 1861–1880 at 1.5, 2, and 4 degrees warming (see Fig. 2), respectively.
Panels (d–f) are similar to panels (a–c) but show the occurrence of heat waves with AT peak ≥ 55 (AT55C).
The USA released the 5th National Climate Assessment in 2023, which presents current and projected climate change and its effects. One set of predictions is the change in number of days above 100 0F ( 38 0C) for different increases in the average global temperature since the pre-industrial revolution time, which is illustrated in Figure \(\PageIndex{5}\) below.
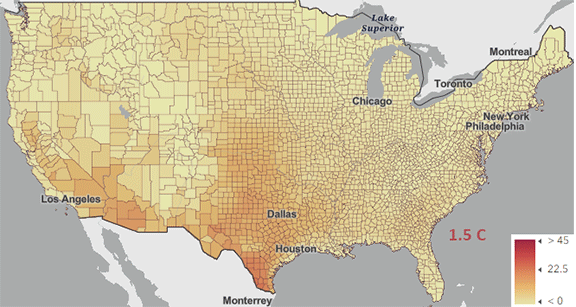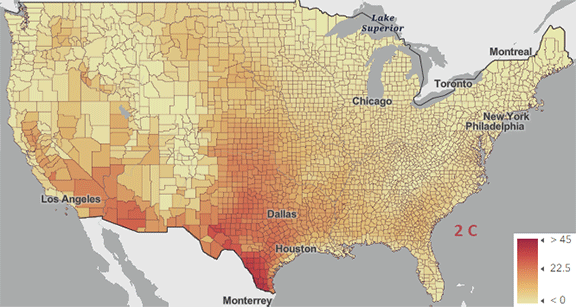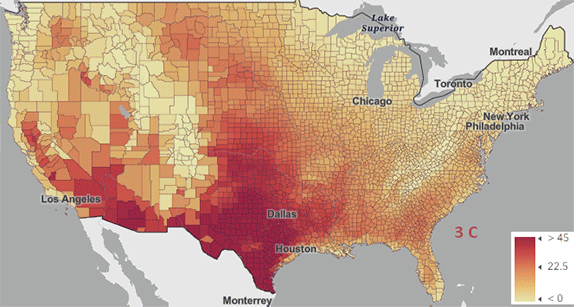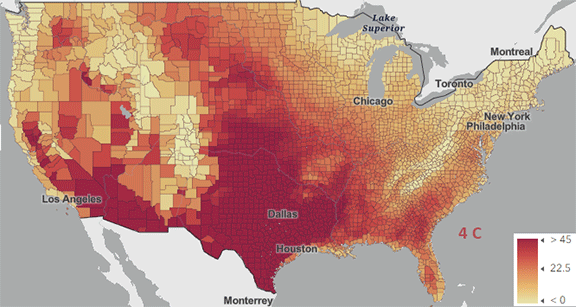
Figure \(\PageIndex{5}\): Change in number of days above 100 0F ( 38 0C) for different increases in the average global temperature since the pre-industrial revolution time. https://www.arcgis.com/apps/mapviewe...3fa3e8bd1fd086
The world reached a net increase of 1.5 0C (2.7 0F) in 2024, shown in the map with the lightest red coloration. The other maps are for 2 0C (3.6 0F), 3 0C (5.4 0F), and 4 0C (7.2 0F). Note the significant change from 1.5 0C (2.7 0F) to 2 0C (3.6 0F).
The USA Weather Service offers a map (still in development) showing the current heat risk.
Heat Exposure and Kidney Disease
Cumulative exposure to high effective temperatures caused by sublethal combinations of heat and humidity leads to chronic kidney disease. This is happening with increasing frequency to workers in poor agricultural areas and others who work in such hot conditions in industry and outdoors. This type of kidney disease is not caused by diabetes, hypertension, or other known glomerular diseases. Chronic kidney disease in such workers was noted in El Salvador and elsewhere in Central America. The disease has a high mortality rate. In El Salvador, the death rate from kidney disease is ten times higher than in the US. Initially, the disease was called Mesoamerican nephropathy.
Biochemical correlates of the diseases are yet unclear. Serum creatinine levels are increased, which might affect renal perfusion and lead to kidney damage. The effects might be generally cumulative or from repetitive episodes of exposure. Sugarcane field workers who report nausea, vomiting, headaches, muscle weakness, back pain, and fevers have high levels of creatinine. Kidney biopsies show inflammation and kidney fibrosis.
Similar diseases in other parts of the world with the same presentation include Sri Lankan nephropathy and Uddanam nephropathy (in the Indian state of Andhra Pradesh). Some have categorized them as CKDu, Chronic Kidney Disease of Unknown Etiology/Uncertain Cause, or chronic kidney disease of non-traditional origin (CKDnt).
Figure \(\PageIndex{6}\) below shows the distribution of kidney disease in the Western Hemisphere in 2019.

Figure \(\PageIndex{6}\): Burden of Kidney Disease. 2019. Men and Women. Pan American Health Organization/WHO.
Other factors, such as increased exposure to herbicides, heavy metals, and microbial agents, might also cause or contribute to the disease. The disease is most prevalent in the hotter regions of affected countries, with a lower incidence in workers at high altitudes. An increased incidence of chronic kidney disease also appears to be occurring in workers in Florida and California.
Biochemical Mechanism for Heat Stroke
The actual biochemical mechanisms of heat stroke effects (circulatory failure, organ injury, uncontrolled clotting, death) are not fully understood. Certainly, cell death plays a significant role, but not via the classical apoptotic pathway, which depends on caspase activation (see Chapter 28.14). Rather, cell death occurs through necroptosis, a caspase-independent pathway. In necroptosis, an upstream protein kinase RIPK3 (receptor-interacting serine/threonine protein kinase 1) activates the effector protein MLKL (mixed lineage kinase domain-like protein) through phosphorylation. Phosphorylated-MLKL then translocates to the cell membrane, leading to calcium influx and plasma membrane damage in the final "execution" phase of cell necrosis.
The activation of RIPK3 and MLKL through other receptors, including the toll-like receptors (TLR3 and TLR4), and tumor necrosis factor receptor 1 (TNF-R1), is shown in Figure \(\PageIndex{7}\) below.

Figure \(\PageIndex{7}\): Activation of RIPK3 by multiple stimuli. Morgan MJ, Kim YS. Roles of RIPK3 in necroptosis, cell signaling, and disease. Exp Mol Med. 2022 Oct;54(10):1695-1704. doi: 10.1038/s12276-022-00868-z. Epub 2022 Oct 12. PMID: 36224345; PMCID: PMC9636380. Creative Commons Attribution 4.0 International License. http://creativecommons.org/licenses/by/4.0/
RIPK3 can be activated by various receptors upon binding their respective ligands. These are TNF receptor 1 (TNF-R1), CD95, death receptors (DR4/5), Toll-like receptors (TLR3/4), and Z-DNA-binding protein-1 (ZBP1)/DAI. In the first three of these pathways (but not TLR3/4 or ZBP1), RIPK1 is required and binds to RIPK3 through its receptor-interacting protein homotypic interaction motif (RHIM). In the case of ZBP1, RIPK3 is recruited directly via the ZBP1 RHIM domain, whereas for TLR3/4, RIPK3 is recruited indirectly via the TRIF RHIM domain. Once activated, RIPK3 autophosphorylates itself, then phosphorylates and activates MLKL, inducing a conformational change and translocation to the membrane, leading to membrane permeabilization. During this process, post-translational modifications positively and negatively regulate the necroptosis pathway. Two E3 ligases, Pellino-1 (PELI1) and carboxy terminus of HSC70-interacting protein (CHIP), may control the basal threshold of necroptosis. Another E3 ubiquitin ligase, TRIM21, is proposed to regulate necroptotic cell death in response to TRAIL. PPM1B suppresses necroptosis by dephosphorylating RIPK3.
The domain structure and phosphorylation sites on human RIPK3 are shown in Figure \(\PageIndex{8a}\) below.

Figure \(\PageIndex{8a}\): Schematic of human RIPK3 domain architecture and the phosphorylation sites identified. Meng, Y., Horne, C.R., Samson, A.L. et al. Human RIPK3 C-lobe phosphorylation is essential for necroptotic signaling. Cell Death Dis 13, 565 (2022). https://doi.org/10.1038/s41419-022-05009-y. Creative Commons Attribution 4.0 International License. http://creativecommons.org/licenses/by/4.0/
Phosphorylation sites with proposed functions are shown at the top. pT224 and pS227 positively regulate necroptosis (green) by recruiting MLKL. pS164 and pT165 negatively regulate necroptosis by inhibiting RIPK3 kinase activity (red). Phosphorylation of T182 (grey) was proposed to promote RIPK3 kinase activity and to recruit PELI1 to mediate proteasomal degradation of RIPK3. Phosphorylation sites with unknown functions are shown on the bottom (white). Asterisks (*) denote multiple serine/threonine on the same peptide, as the exact phosphorylation site could not be unambiguously identified.
Figure \(\PageIndex{8b}\) shows necrosome, a complex containing multiple activated RIPK3s and RIPK1. Aggregation of RIPK3 occurs through the RHIM (RIP homotypic interaction motifs) domain through the formation of amyloid fibers. The necrosome then phosphorylates MLKL, which forms oligomers and traffics to the membrane.

Figure \(\PageIndex{8b}\): A schematic diagram of the current model of necroptosis signaling. Meng, Y. et al., ibid.
RIPK3 kinase activity is negatively regulated by pS164/pT165, which promotes apoptotic pathways. Under basal conditions, in the absence of inhibitory pS164/pT165, RIPK3 autophosphorylates T224/S227 in a MLKL-dependent manner, allowing MLKL to be recruited to the kinase domain of RIPK3. Upon TNF signaling in the absence of cIAPs and Caspase-8 activity, MLKL is phosphorylated in the necrosome before disengaging from the RIPK3 kinase domain, undergoing a structural transition, and assembling into active oligomers, which are then trafficked to the plasma membrane, where they accumulate as hotspots to enact cell death.
Figure 8b above also shows that an internal viral DNA sensor protein can activate RIPK3. That protein is ZBP1, or Z-DNA-Binding Protein 1, which also binds Z-RNA. Nuclear Z-RNA can be derived from viruses like influenza A, leading to activation of the same pathway. Cytokine expression then produces a systemic inflammatory response.
In addition to apoptosis and necroptosis, another type of programmed cell death caused by inflammation is called pyroptosis. Usually occurring in macrophages infected with bacteria, pyroptosis activates intracellular inflammasomes, leading to the production of inflammatory cytokines via caspase-mediated proteolysis. In pyroptosis, proteins called gasdermins are cleaved by caspases, and their N-terminal sections self-assemble at the cell membrane to form pores, releasing the inflammatory cytokines IL-1β and IL-18.
A final programmed cell death pathway for virally infected cells is called PANoptosis, which uses the PANoptosome complex and has downstream results not explained by the other three programmed cell death pathways (pyroptosis, apoptosis, and necroptosis). ZBP-1 leads to the activation of RIPK3, caspase-8 (a key player in the apoptosis pathway), and the NLRP3 inflammasome.
ZBP-1 seems to play a key role in heat stroke. Its concentration increases under heat stress, mediated by the heat shock transcription factor 1 (HSF1), which is induced by cellular stress. HSF1 induces a heat shock response, leading to increased transcription of chaperones and heat shock proteins (HSPs), such as ZBP-1. Deletion/inactivation of ZBP-1, RIPK3, MLKL, or caspase 8 reduces heat stroke. The main role of ZBP-1 in cell death from heat stroke arises from the RIPK3/MLKL pathway and, to a lesser extent, through cross-talk with the classical apoptosis pathway through caspase 8.
How does ZBP-1 activate cell death during heat stroke without binding to and activating dsDNA or RNA derived from a viral infection? Does ZBP-1 have an endogenous ligand other than viral Z-RNA or Z-DNA? First, explore the domain structures of key proteins in the RIPK3 activation pathway. Figure 1 above shows three key proteins, RIPK1, TRIFF, and ZBP1, that interact with RIPK3. These proteins, including RIPK3, have an RHIM domain that mediates protein-protein interactions. Figure \(\PageIndex{9}\) shows the domain structure of our key protein, ZBP-1, the cytosolic Z-DNA/Z-RNA sensor.
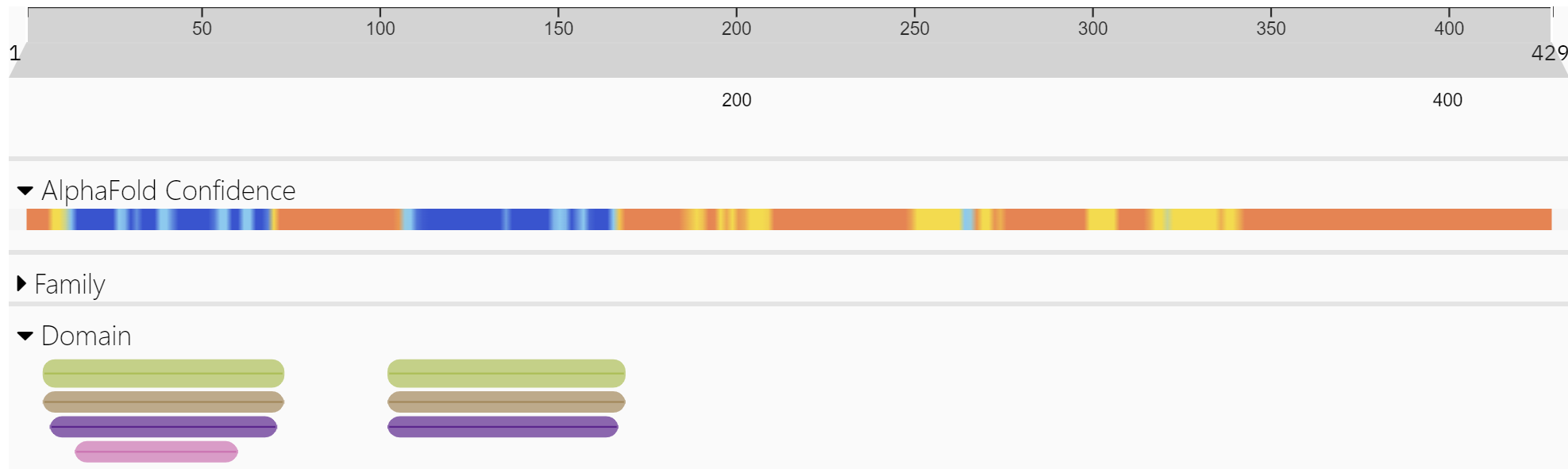
Figure \(\PageIndex{9}\): Domain structure of ZBP-1 (http://www.ebi.ac.uk/interpro/protein/UniProt/Q9H171/ )
The green bars in the N-terminal part of the protein are the Z-DNA binding domain. These are also called Zα domains. These regions are the most ordered in the protein, as indicated by the blue in the AlphaFold confidence bar.
Figure \(\PageIndex{10}\) shows an interactive iCn3D models of the AlphaFold-predicted model of human Z-DNA-Binding Protein 1 (ZBP1), (Q9H171)
-_(Q9H171.png?revision=1&size=bestfit&width=568&height=270)
Figure \(\PageIndex{10}\): AlphaFold-predicted model of human Z-DNA-Binding Protein 1 (ZBP1), (Q9H171). (Copyright; author via source). Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...FMCRhJwHFVb1X7
The spacefill atoms labeled M1 represent the N-terminal methionine of the protein. The two Z-DNA binding domains are well-ordered and are shown as blue cartoons. Much of the protein can't be predicted as it is most likely intrinsically disordered. Two fairly well-structured motifs, shown in magenta and cyan, are the RHIM1 and RHIM2 protein interaction motifs, which can self-associate via their amyloid-like structures. These motifs allow ZBP1 to bind to other proteins with RHIM motifs, leading to cell death via necrosis. The C-terminal domain appears to be involved in type I interferon-mediated signal transduction in response to DNA.
Figure \(\PageIndex{11}\) shows an interactive iCn3D model of the second Z-DNA binding domain of human DAI (ZBP1) in complex with Z-DNA (3EYI)
_in_complex_with_Z-DNA_(3EYI)V3.png?revision=1&size=bestfit&width=649&height=236)
Figure \(\PageIndex{11}\): Second Z-DNA binding domain of human DAI (ZBP1) in complex with Z-DNA (3EYI). (Copyright; author via source). Click the image for a popup or use this external link: https://structure.ncbi.nlm.nih.gov/i...SLBmo2xqV1WpS6
Without viral DNA or RNA, the Zα domain can bind endogenous ligands. Moreover, a deficiency in RIPK1 or RHIM in RIPK1 also triggers ZBP1-mediated necroptosis and inflammation, and its Zα domain is required. If nuclear export is stopped, ZBP1 activates nuclear RIPK3 and then necroptosis. This suggests that nuclear ZBP1 interacts with endogenous nuclear Z-nucleic acids, likely Z-RNA from retroelements, to activate RIPK3-dependent necroptosis and may contribute to some forms of chronic inflammation.
Here are a series of findings on RIPK3-dependent cell death on heat stress in mouse fibroblasts that show that Z-nucleic acid binding to ZBP1 is not required for heat stress effects:
- Heat (43°C for 2 hr) induces phosphorylation of RIPK3 and MLKL within 2 hours and cleavage of pro-caspases and GSDME within 6 hours, but none occurs if RIPK3 is deleted.
- Deletion of ZBP1, but not RIPK1, affects heat-induced death, indicating that heat stress acts through ZBP1 and RIPK3.
- In ZBP1-deficient mice, the effects of heat stress (clotting, inflammation, organ injury, and death) were prevented.
- Mutations in the RHIM domain, but not the Zα domains (made to prevent Z-nucleic acid binding) or in the C-terminal signaling region (to stop signaling), prevented death from heat stress. Hence, Z-nucleic acid binding is not required, but it may contribute to heat-stress-induced cell death.
- Heat stress caused the aggregation of a ZBP1-GFP (green fluorescent protein) fusion protein through the RHIM domains of ZBP-1.
Hence, ZBP1 is an innate pathogen sensor and an initiator of heat-related death in the absence of pathogens.
Heat Stroke-Induced Epigenetic Changes
Short of death, heat stroke can also cause long-term health issues. Increasing global temperatures are forcing people to work in more dangerous temperatures and at night to reduce heat exposure. Data suggest that people with heat-related illnesses are more susceptible to health consequences from additional heat exposure. This has been noted in exertional heat illnesses. (such as in athletes). Additional long-term effects on immune regulation have been observed. Epigenetics may play a role in long-term effects such as greater vulnerability to additional heat challenges. Studies show that a single episode of exceptional heat stroke changes DNA methylation patterns in bone marrow-derived monocytes from mice. Monocytes become immunosuppressed, leading to increased microbial disease and reduced heat shock responses. Epigenetic changes are passed on to progeny monocytes, leading to compromised function. The epigenetic changes persist for 30 days or more, and we noted them in inflammatory cell signaling pathways. This suggests a mechanism for the reduced tolerance to those with previous heat-related illnesses.
Cardiovascular, Pulmonary Diseases and Cancer
Many factors can cause these diseases and lead to mortality. For example, cancer mortality can increase due to the lower availability of health care arising from extreme weather disasters that impact health facilities and access to them. Early detection of many cancers is key to survival. Instead of discussing climate change's links to cancer, we will focus on pollution, particularly small particles.
PM2.5 particulate pollution effects on cardiovascular and pulmonary health
Pollution from the combustion of fossil fuels contributes to many chronic diseases. Here, we will focus on one type of pollutant, particulate matter, which can be inhaled. These particles can be liquids, solids, or combinations of both. They are classified according to size with common categories, including:
- PM10: diameters < 10 uM = 10,000 nm;
- PM2.5: fine particles with diameters < 2.5 um = 2500 nm
- PM0.1: ultra-fine particles with diameters < 0.1 μm = 100 nm (also called nanoscopic particulate matter or NPM)
Figure \(\PageIndex{12}\) below shows the relative sizes of PM10 and PM2.5 particles compared to other biological structures.

Figure \(\PageIndex{12}\): Relative sizes of PM particles compared to biological structures. Sotirios Papathanasiou. Particulate Matter (PM2.5) Mega Guide. With Permission. https://seetheair.org/2022/05/16/par...-5-mega-guide/
Figure \(\PageIndex{13}\) below shows the relative sizes of PM0.1 particles compared to a PM2.5 particle.

Figure \(\PageIndex{13}\): Relative sizes of PM01 particles compared to a PM2.5 particle.Sotirios Papathanasiou, ibid.
Composition of PM2.5 particles
PM2.5 particles collected from polluted city air can be purchased from the National Institute of Standards and Technology (NIST, SRM 1648a) and used in experimental studies of living cells. It is typically added to water, and a suspension is produced through sonication. PM2.5 particles are derived from human sources, such as vehicle and industrial emissions, and from natural processes, such as biomass burning and windblown dust. They can also include salts from land and ocean sources.
They arise from burning fossil fuels and the wear and tear of products such as automobiles (including tires). PM2.5 particles contain mainly black carbon, polycyclic aromatic hydrocarbons (PAH), aryl hydrocarbons, volatile organic compounds (VOCs), as well as minerals, ions (sulfate, nitrate, ammonium), and general biological materials. The metal composition includes Group 1A (K, Na, Fr), Group 2A (Ca, Mg), Group 3A (Al), transition metals (Al, As, Cr, Fr, Mn, Pb, Ti, Zn), and counter ions Br and Cl. They also contain silicon and silicates. Of course, particles in the air, including dust, also derive from non-anthropogenic sources. Atmospheric dust is also produced from land by winds and volcanic eruptions. In homes, dust contains a high concentration of dead skin cells. along with pollens, hair, fur, and fibers from clothes and paper. Humans have evolved with particulates in the air, but large increases in their abundance, driven by human activities in many parts of the world, pose serious health consequences.
In addition, reactions among atmospheric pollutants produce "secondary" pollutants. One, the tropospheric gas ozone (O3), produced by hydrocarbons and nitrogen oxides, is a known health risk, and its levels are higher in cities on sunny, hot, and humid days. Secondary organic carbon (SOC) is generated from primary organic carbon, typically volatile organic compounds (VOCs), through photochemical oxidation. These VOCs (like m-xylene and 1,2,4-trimethylbenzene) can produce aerosols (larger particles, called secondary organic aerosols by reacting with each other to produce larger structures. Terpenes containing isoprene units like α-pinene and limonene (a monoterpene found in large abundance in fruit peels) are reactants for the products of much larger structures. (See Chapter 10.1 and Chapter 21.6 for a review of isoprene and terpenes). Figure \(\PageIndex{14}\) below shows generalized pathways for forming particulate SOCs from smaller terpenes.

Figure \(\PageIndex{14}\): Proposed mechanisms for the formation of C20H33N3O12, C20H32N4O14, and C30H48N4O16 in the simultaneous oxidation (MIX) experiment. Takeuchi, M., Berkemeier, T., Eris, G. et al. Non-linear effects of secondary organic aerosol formation and properties in multi-precursor systems. Nat Commun 13, 7883 (2022). https://doi.org/10.1038/s41467-022-35546-1. Creative Commons Attribution 4.0 International License. http://creativecommons.org/licenses/by/4.0/.
The gas-phase mechanism via cross-reactions of α-pinene and limonene peroxy radicals (RO2·), and the particle-phase mechanism via hemiacetal formation involving α-pinene and limonene oxidation products. APN-RO2, APN(=O), and APN(-OH) represent α-pinene oxidation intermediates (i.e., RO2·), α-pinene oxidation products containing carbonyl groups, and α-pinene oxidation products containing hydroxyl groups, respectively.
Figure \(\PageIndex{15}\) below shows the morphology of PM2.5 particles.

Figure \(\PageIndex{15}\): Morphology of PM2.5 particles. The scale bars are 20 μm for image (a) 2 μm for image (b) 1 μm for image (c) 40 μm for image (d). Shi, Y., Ji, Y., Sun, H. et al. Nanoscale characterization of PM2.5 airborne pollutants reveals high adhesiveness and aggregation capability of soot particles. Sci Rep 5, 11232 (2015). https://doi.org/10.1038/srep11232. Creative Commons Attribution 4.0 International License. http://creativecommons.org/licenses/by/4.0/.
Panel (a) shows a large area image of collected PM2.5 on the filamentary filter.
Panels (b) and (c) show SEM images of particles with flat and rough top surfaces, respectively.
Panel (d) shows an SEM image of the PM2.5 transferred to a silicon substrate. Inset, zoom-in SEM image of an Iron-rich particle.
Many PM2.5 particles have rough surfaces that can deform and interact more readily with (i.e., stick to) other particles through noncovalent interactions, forming even larger particles.
Figure \(\PageIndex{16}\) below shows the elemental composition of rough, semi-rough, and flat PM2.5 particles.

Figure \(\PageIndex{16}\): EDAX Chemical composition histogram of the particles collected with SEM/EDAX classified by surface roughness; a larger surface roughness (and therefore, stickiness and deformation) is linked to a larger content of carbon while particles with a flat surface (low stickiness and viscosity) are richer in oxygen and metals. Shi, Y. et al., ibid.
Figure \(\PageIndex{17}\) below shows electron micrographs of actual airborne particles. Most are PM2.5 particles with diameters < 2.5 uM = 2500 nm.

Figure \(\PageIndex{17}\): A collage of SEM images for airborne particulates. (A) General classification of airborne particulates: (B) particulates with seed-coating composite morphology; (C) sulfate particulates with different morphologies. Clara Yuan Li et al, Journal of Environmental Protection, Vol.7 No.10, 2016. https://www.scirp.org/journal/paperi...?paperid=71021. Creative Commons Attribution 4.0 International License.
Given their composition and structures, it doesn't take much thought to realize that the particles must cause significant health effects. Would you want to breathe these particles routinely?
Health Effects of PM2.5 Particles
PM2.5 particles are associated with many types of illness, including cardiovascular and pulmonary diseases (including asthma) and cancer. Since they are small, they can be easily inhaled and deposited in the lung alveoli, where they can enter the bloodstream and be deposited in tissues. Particles up to 240 nm (0.25 μm) can cross the placenta, and black carbon particles have been found to cross the placenta. PM2.5 particles can cause inflammation, DNA damage, and organelle dysfunction, and generate free radicals, most likely involved in these toxic health effects.
Aerosol particles can even have acute and lethal effects. During the Great London Smog in December 1952, around 12,000 people died in two weeks from its effects. Figure \(\PageIndex{1}\) shows people in the thick smog from that event. The smog consisted of acidified water droplets from SO2 and NO2 released by burning coal containing sulfur. This gas can be oxidized to sulfate in gas-phase reactions, probably through the .OH free radical or in aqueous phase reactions using O3, peroxides, and NO2 as reactants/catalysts.
The reaction of SO2 with NO2 in aqueous droplets is shown below:
\begin{equation}
\mathrm{SO}_2(\mathrm{~g})+2 \mathrm{NO}_2(\mathrm{~g})+2 \mathrm{H}_2 \mathrm{O}(\mathrm{aq}) \rightarrow 2 \mathrm{H}^{+}(\mathrm{aq})+\mathrm{SO}_4^{2-}(\mathrm{aq})+2 \mathrm{HONO}(\mathrm{g})
\end{equation}
Given the stoichiometry of the reaction with NO2, the reaction proceeds significantly only in the presence of high NO2. The reaction is favored under high relative humidity. In large water droplets in clouds, the droplets are not very acidic, but as water evaporates from them, the sulfate concentration and acidity increase dramatically. However, the oxidation rate and solubility decrease as the acidity increases.
Major cities in China, including Beijing and Xi'an, have experienced high levels of haze and particulate matter in the atmosphere until recently. Yet these smogs were not as lethal (very acidic) as those in London because NH3 added to the droplets neutralized the acidic particles. Atmospheric ammonia is derived from large amounts of agricultural fertilizers that are aerosolized, from vehicles that produce NH3 in catalytic converters, and from urea used in the catalytic reduction in diesel engines. The relevant production of sulfates in the presence of NH3 is shown in the equation below.
\begin{equation}
\begin{aligned}
2 \mathrm{NH}_3(\mathrm{~g})+\mathrm{SO}_2(\mathrm{~g})+2 \mathrm{NO}_2(\mathrm{~g}) & +2 \mathrm{H}_2 \mathrm{O}(\mathrm{aq}) \rightarrow 2 \mathrm{NH}_4^{+}(\mathrm{aq}) + \mathrm{SO}_4^{2-}(\mathrm{aq})+2 \mathrm{HONO}(\mathrm{g})
\end{aligned}
\end{equation}
Figure \(\PageIndex{18}\) below shows a ghostly image of pedestrians in London during the Great Fog.

Figure \(\PageIndex{18}\): Ghost-like pedestrians going through the smog. https://heritagecalling.com/2022/12/...f-london-1952/. Public Domain
The prevailing weather conditions (cold temperatures) increased emissions from coal use. A stalled high-pressure system and resulting low winds led to stagnant air, allowing increasingly acidic PMs to build up. The appalling death toll led politicians to pass the Clean Air Act in 1954, which, over many years, led to huge improvements in air quality in London and dramatically reduced negative health effects and deaths. This was a prelude to the US Clean Air Act, which dramatically improved air quality.
High levels of PM2.5 remain prevalent worldwide, though there have been dramatic decreases in the US. The notable exceptions occur during forest fires that are exacerbated by climate change. In the US, the Air Quality Index (AQI) is one indicator of health risk. It measures the value of 5 pollutants: fine particles (PM2.5 and PM10), ground-level ozone, SO2, NO2, and CO. The AQI at a given time is determined by the highest pollutant. In haze from smoke, the reported AQI reflects PM2.5 particles. AQI values < 50 represent good air quality, while an AQI value over 300 represents hazardous air quality. Western forest fires in Oregon in September 2020 led to an AQI of 611 in Madras, Oregon. Forest fires in Eastern Canada and a slow-moving weather system temporarily led to PM2.5 levels over 800 (mg/m3) in New York City on June 7, 2023. No place is immune to PM2.5 particles from wildfires and human-caused pollution.
Severe forest fires in Canada in 2023 led to significant health consequences in the US. On 19 smoke days (Air Quality Index ≥101, which is unhealthy for sensitive groups) between April 30–August 4, 2023), there was a 17% increase in visits to emergency departments for asthma-associated conditions. In counties affected by California wildfires in 2015-2017, there was a large increase in "out-of-the-hospital coronary arrest". In the first 3 months after the 2025 fires in Los Angeles, there was a 46% increase in heart-attack-related emergency room visits at Cedars-Sinai Hospital.
- AirNow - gives present pollution data, primarily PM2.5 levels, based on US zip codes.
The following interactive graphs show the changes in PM2.5 particles over time (from Hannah Ritchie and Max Roser (2019), OurWorldInData.org/outdoor-air-pollution • CC BY. Source: Brauer et al. (2017) via World Bank).
Figure \(\PageIndex{19}\) shows the share of the population exposed to PM2.5 levels higher than the World Health Organization's suggestions.
Figure \(\PageIndex{19}\): Share of the population exposed to PM2.5 levels higher than those suggested by the World Health Organization.OurWorldInData.org/outdoor-air-pollution • CC BY. Source: Brauer et al. (2017) via World Bank
Figure \(\PageIndex{20}\) below shows the death rate from PM2.5/100,000 people in 2017 in countries worldwide.
Figure \(\PageIndex{20}\): Death rate from PM2.5/100,000 people in 2017 in countries worldwide.
Recent estimates based on WHO guidelines suggest that, in India, an average of 16.6 million annual deaths (13-22 million) from 2009 to 2019 were attributable to PM2.5 (about 25% of total mortality), far above the estimates shown in the graph above.
Mechanisms for PM2.5 Health Effects
In Chapter 5.4, we discussed how solids such as silica, cholesterol crystals, uric acid crystals, and even aggregated proteins such as prions can be engulfed by monocytes/macrophages (much as they engulf bacteria as part of their immune function) in a process called phagocytosis. The particles are enveloped in a plasma bilayer-derived membrane, which buds into the cell. This vesicle merges with a lysosome, which gets damaged in the process. They then release ATP into the cytoplasm, which acts as a damage signal, activating inflammation.
PM2.5 particles can also be taken up by phagocytosis to produce intracellular phagosomes. Pinocytosis and caveolin/clathrin-mediated endocytosis can also take them up. The resulting endosome fuses with lysosomes and mitochondria, causing damage. The heavy metals from PM2.5 particles released into the cell also contribute to damage.
Smaller particles with diameters less than 0.1 μm (100 nM), sometimes called nanoscale particulate matter (NPM), offer large surface areas for protein adsorption. The adsorbed proteins form a "crown" called a protein corona. The corona is larger than the NPM. Proteins bound to the particles include hemoglobin, albumin, and fibrinogen. The corona also mediates cellular interactions and participates in the mechanisms that lead to inflammation and cellular dysfunction. Because the extracellular matrix includes proteins such as collagen and fibrin, the interaction of PM2.5 with fibrin has been studied as a model for how particles might interact with cells. In particular, lung fibroblasts embedded in a 3D fibrin matrix (i.e., a 3D organotypic culture) were exposed to NPM particles with protein coronas, and the effects of these particles on cell proliferation, oxidative stress, and other parameters were monitored. Figure \(\PageIndex{21}\ below characterizes the interaction of the NPMs with the fibroblast in the 3D culture.

Figure \(\PageIndex{21}\): Physicochemical characterization of airborne particulate matter. Li, Y., Wang, P., Hu, C. et al. Protein corona of airborne nanoscale PM2.5 induces aberrant proliferation of human lung fibroblasts based on a 3D organotypic culture. Sci Rep 8, 1939 (2018). https://doi.org/10.1038/s41598-018-20445-7. Creative Commons Attribution 4.0 International License. http://creativecommons.org/licenses/by/4.0/.
Panel (A) shows SEM images of airborne PM2.5 (nanoscale PM2.5 is marked in red).
Panel (B) shows an EDX (Energy-dispersive X-ray spectroscopy) analysis of PM2.5.
Panel (C) shows an atomic force microscopy (AFM) image of nanoscale PM2.5 (NPM) from air pollutant samples (scale 3.5 μm).
Panel (D) AFM image of airborne NPM from air pollutant samples (scale 1 μm).
Panel (E) shows an SEM image of airborne NPM from air pollutant samples.
Panel (F) shows an SEM image of an NPM-protein corona.
Panel (G) shows a schematic diagram of the biological interaction between NPM and the protein.
Panel (H) shows an FTIR spectrum of NPM, serum, and NPM-protein corona.
The NPM-protein corona particle leads to the proliferation of 3D-cultured human lung fibroblast cells beyond stimulation with NPMs or serum alone. The bigger the size of the corona, the greater the proliferative effect. This is consistent with the extensive lung fibrosis observed after chronic exposure to PM2.5 particles. Reactive oxygen species also increased in the presence of NPM-corona particles. These data suggest that the NPM-protein corona is important in PM2.5-induced lung fibrosis and pulmonary disease.
Neural Effects of PMs
PM particles, particularly the ultrafine PM0.1 particles (UFP), can enter the brain and affect neural function. Exposure to PM2.5 over long periods is associated with an increased incidence of dementia and Alzheimer's Disease (AD). Four particular components of PMs (SO42−, NH4+, black carbon, and organic matter) were most associated with a higher risk of dementia and AD. In the US, the first admission to a hospital for Parkinson's, Alzheimer's, and other dementias is "significantly" associated with the average annual mean PM2.5 exposure.
Even low levels of exposure pose risks. Transgenic mice expressing mutant forms of human presenilin 1, amyloid precursor protein, and tau were exposed to subchronic, "real-world" levels of PM2.5 via inhalation for 3 months. Neuronal loss was observed in the cortex, but no motor or cognitive impairment was noted. Increased levels of phosphorylated Tau and free radical formation, as evidenced by malondialdehyde (a marker of oxidative stress), were observed in the hippocampus and olfactory centers, consistent with inhalation of PM2.5 through the noise. No abnormal amyloid plaques were observed in this short exposure time.
PM-induced neuronal damage occurs through the generation of reactive oxygen species, increased inflammatory responses, and organelle damage (all of which are interrelated). Even in skin cells (keratinocytes), exposure to PM2.5 particles led to increased ROS and malondialdehyde levels, decreased superoxide dismutase levels, and increased DNA damage. Inflammatory Caspase levels also increased.
Cells have mechanisms to detect and eliminate aberrant species before they cause more severe biological effects. One process is autophagy, which degrades misfolded proteins and damaged organelles, both of which are important for normal neural function. Aberrant autophagy is a key player in the pathogenesis of dementia. A second pathway is ferroptosis, an apoptotic pathway that kills cells that have accumulated large amounts of iron ions, which are themselves free radicals. Through the Fenton reaction and other mechanisms, iron ions can generate damaging reactive oxygen species and oxidize lipids, proteins, and nucleic acids (see Chapter 12.3).
Several key proteins are involved in antioxidant defense and autophagy:
- NRF2 (Nuclear factor erythroid 2-related factor 2): This transcription factor binds to antioxidant response elements (ARE) in protective genes.
- Keap1 (Kelch-like ECH-associated protein 1): An adapter protein that targets NRF2 for ubiquitination and, as such, is a sensor for oxidative stress. Under those conditions, electrophilic metabolites post-translationally modify reactive Cys side chains, inactivating ubiquitin ligase activity. This increases NRF2 and subsequent transcription of antioxidant genes.
- SQSTM1 aka p62 (Sequestosome-1): This protein bridges autophagosomes and polyubiquitinated proteins (cargo for degradation).
Hence, under low oxidative stress, NRF2 is degraded by KEAP1, keeping its concentration low. If SQSTM1 (p62), increases, and reduced autophagy, p62 biding to the NRF2 sites on KEAP1, leading to the release of NRF2, its transfer to the nucleus, and activation of gene transcription of protective genes. These three proteins also protect cells from ferroptosis. In the long term, neural cell death occurs during dementia. Hence, apoptosis, necroptosis, and pyroptosis (from activation of the inflammatory response mediators caspase, Gasdermin, and key cytokines like IL-1β and IL-18) do "win out" to eventually kill cells.
Figure \(\PageIndex{22}\) below shows the domain structure of SQSTM1 (panel A) and the signaling process described above.

Figure \(\PageIndex{22}\): Positive feedback-loop of Nrf2 activation by p62/SQSTM1. Vomund S, Schäfer A, Parnham MJ, Brüne B, von Knethen A. Nrf2, the Master Regulator of Anti-Oxidative Responses. Int J Mol Sci. 2017 Dec 20;18(12):2772. doi: 10.3390/ijms18122772. PMID: 29261130; PMCID: PMC5751370. Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Panel (A) shows the domain structure of p62/SQSTM1;
Panel (B) shows p62/SQSTM1 as an important protein in selective autophagy, as it binds to Keap1 and other long-lived proteins, forming polyubiquitinated protein aggregates. Furthermore, it binds to the autophagy marker LC3 within the autophagosome, thereby leading the aggregated proteins into the autophagosome. After fusion with a lysosome, proteins and organelles, such as mitochondria, are degraded within the autophagosome. By binding to Keap1, p62/SQSTM1 stabilizes Nrf2 and enhances its nuclear translocation, leading to the activation of Nrf2 target genes (↑ = upregulation of Nrf2 target genes). One of these genes is p62/SQSTM1
PM2.5 particles interfere with the protective autophagy and ferroptosis pathways, leading to increased NRF2 activity and the expression of antioxidant genes. These processes are beneficial for ridding cells of aberrant particles and killing damaged cells in normal conditions, but not when neuronal cells are exposed to PM2.5 particles.
Lysosomal membrane permeabilization (LMP), as well as mitochondrial and ER damage, may be a likely mechanism for initiating ultimate neuronal death on long-term exposure to PM2.5 particles. PM2.5 particles inhibit lysosomal activity and increase permeability, releasing degradative enzymes into the cytoplasm. Ultimately, increased or decreased Nrf2 activation leads to disease states.
PMs and Cancer
Long-term exposure can also cause lung cancer. This is associated with the conversion of lung cells from normal epithelial to mesenchymal cells (the EMT transition). Phenotypically, mesenchymal cells can migrate, invade other tissues, and cause enhanced extracellular matrix production, all hallmarks of tumor cells. This EMT transition is associated with significant changes in cell signaling and transcription factor production. These changes are documented in Figure \(\PageIndex{23}\) below and its caption.

Figure \(\PageIndex{23}\): Brief schema of the putative signaling transduction mechanisms underlying EMT. Xu et al., Front. Physiol., 29 November 2019. Sec. Renal Physiology and Pathophysiology. Volume 10 - 2019 | https://doi.org/10.3389/fphys.2019.01404. Creative Commons Attribution License (CC BY)
Activation of the Wnt/β-catenin, PI3K/Akt, Ras/ERK, TGF-β/SMAD2/3, BMP/SMAD1/5/8, JAK/STAT3, Shh, and Notch pathways is highly correlated with EMT. After ligand-receptor binding, intracellular secondary messengers are activated and initiate downstream transduction, which generally induces the nuclear translocation of signaling-specific TFs and the transcriptional regulation of EMT-related genes, such as CDH1 and CDH2, EMT TFs, and mesenchymal markers, accompanied by a series of alterations in cellular physiological or pathological activities (e.g., dysjunction of adherin junctions, cytoskeleton remodeling, and increase of cellular motility). Arrows represent the molecular interactions in which downstream messengers are activated; T-shaped arrows represent inhibitive molecular interactions. EMT, epithelial-mesenchymal transition; PI3K, phosphoinositide 3-kinase; ERK, extracellular signal-regulated protein kinase; TGF-β, transforming growth factor β; JAK, Janus kinase; Shh, sonic hedgehog; TFs, transcription factors.
PM2.5 particles, their deleterious contents (heavy metal ions, PAHs, etc.), and the ROS generated are associated with the changes seen in the EMT transition. These include activation of transforming growth factor β (TGF-β)/SMADs, NF-κB, growth factor (GF)/extracellular signal-regulated protein kinase (ERK), GF/phosphatidylinositol 3-kinase (PI3K)/Akt, wingless/integrated (Wnt)/β-catenin, Notch, Hedgehog, high mobility group box B1 (HMGB1)-receptor for advanced glycation end-products (RAGE), and aryl hydrocarbon receptor (AHR) signaling cascades, and to cytoskeleton rearrangement.
Summary
(Summary written by Claude, Anthropic)
This chapter examines how climate change and fossil fuel combustion damage human health at the molecular level, organized around the One Health framework — the recognition that human, animal, plant, and environmental health are inseparably linked. Two major topics are covered: heat-related illness and particulate matter (PM) pollution.
Heat-Related Illness and Kidney Disease. Heat waves are increasing in frequency, duration, and severity globally. Major events — the 2003 European heat wave (70,000 deaths), the 2010 Russian heat wave (55,000 deaths), and the 2022 European heat wave (60,000 deaths) — establish extreme heat as a leading cause of climate-attributable mortality, with a clear linear relationship between rising summer mean temperatures and heat-related deaths. The elderly and women are disproportionately affected. Physiologically, heat imposes cardiovascular strain through sustained heart rate elevation; under humid conditions this develops at temperatures as low as 34°C. US National Climate Assessment projections show a dramatic increase in days above 100°F between 1.5°C and 2°C of warming — a threshold the world crossed in 2024 — with the difference between these two warming scenarios being especially stark.
Cumulative occupational heat exposure also causes CKDu (chronic kidney disease of uncertain etiology), first identified as Mesoamerican nephropathy in Central American sugarcane workers and subsequently recognized in Sri Lanka, India, and now among agricultural workers in Florida and California. CKDu presents with elevated serum creatinine, renal inflammation, and fibrosis in the absence of traditional risk factors (diabetes, hypertension, glomerular disease), correlates strongly with regional heat exposure, and carries high mortality rates — ten times higher than in the US in El Salvador.
Biochemical Mechanisms of Heat Stroke. Heat stroke kills cells primarily through necroptosis rather than classical apoptosis. The central pathway runs through RIPK3, which first autophosphorylates (at pT224 and pS227) and then phosphorylates MLKL; phosphorylated MLKL undergoes a conformational change, oligomerizes, traffics to the plasma membrane, and causes membrane permeabilization, calcium influx, and cell lysis. Negative regulatory phosphorylation of RIPK3 at pS164 and pT165 diverts signaling toward apoptosis. Multiple activated RIPK3 molecules assemble via their RHIM domains into amyloid-like necrosome complexes that amplify MLKL phosphorylation. E3 ligases PELI1 and TRIM21 regulate pathway thresholds.
Under heat stress, ZBP1 (Z-DNA Binding Protein 1) is upregulated by the heat shock transcription factor HSF1 and activates RIPK3 through its RHIM domain — not its Zα nucleic acid-binding domain. This is mechanistically distinct from viral Z-RNA sensing: mutations in ZBP1's RHIM domain prevent heat-stroke pathology, while Zα domain mutations do not. ZBP1 aggregates through its RHIM domains under heat stress, driving RIPK3 activation in the absence of any pathogen. Mouse knockouts of ZBP1, RIPK3, or MLKL dramatically reduce heat-stroke clotting, inflammation, organ injury, and death. The chapter also introduces pyroptosis (gasdermin cleavage by caspases produces membrane pores releasing IL-1β and IL-18) and PANoptosis (a convergent death program integrating necroptosis, apoptosis, and pyroptosis via ZBP1 and the NLRP3 inflammasome) as additional heat injury mechanisms.
Beyond acute cell death, heat stroke induces lasting epigenetic changes: a single episode alters DNA methylation patterns in bone marrow monocytes, causing immunosuppression and reduced heat shock responses that persist for 30+ days and are inherited by daughter cells. This provides a molecular basis for the clinically observed reduced heat tolerance in individuals with prior heat illness.
PM2.5: Sources, Composition, and Health Effects. PM2.5 particles (≤2.5 μm diameter) from fossil fuel combustion carry a complex toxic cargo: black carbon, polycyclic aromatic hydrocarbons, transition metals (Mn, Pb, Zn, Fe, Cr, As), sulfate, nitrate, ammonium, and VOCs. Secondary atmospheric reactions among these pollutants generate additional hazards — tropospheric ozone from NOx/hydrocarbon reactions, and secondary organic aerosols from terpene (α-pinene, limonene) photochemical oxidation. The Great London Fog of 1952 — which killed ~12,000 people in two weeks from highly acidic sulfate aerosols formed by SO₂ oxidation in aqueous droplets catalyzed by NO₂ — established the lethal potential of PM pollution and directly prompted the UK Clean Air Act. Today, wildfire smoke (worsened by climate change) creates acute PM2.5 spikes of global reach, with Canadian fires driving AQI readings over 800 in New York City in 2023 and the 2025 Los Angeles fires producing a 46% spike in cardiac emergency visits.
PM2.5 particles enter cells via phagocytosis, pinocytosis, and clathrin/caveolin-mediated endocytosis. Nanoscale PM0.1 particles acquire a protein corona — adsorbing hemoglobin, albumin, and fibrinogen — that amplifies inflammatory signaling and drives lung fibroblast proliferation in 3D culture models, consistent with the extensive pulmonary fibrosis seen in chronic PM2.5 exposure. The primary toxicity mechanisms are oxidative stress and inflammation. The transcription factor NRF2, normally targeted for ubiquitination by Keap1, is released when electrophilic PM2.5 metabolites modify Keap1 cysteine residues, upregulating antioxidant genes. The scaffold protein p62/SQSTM1 bridges autophagosomes to polyubiquitinated cargo and creates a positive feedback loop by displacing NRF2 from Keap1 when autophagy is overwhelmed. When these defenses fail, lysosomal membrane permeabilization releases digestive enzymes into the cytoplasm, and mitochondrial and ER damage trigger apoptosis, necroptosis, and pyroptosis.
In the nervous system, PM0.1 ultrafine particles cross the blood-brain barrier and accumulate in neural tissue. Long-term PM2.5 exposure is significantly associated with Alzheimer's disease, Parkinson's disease, and other dementias. In transgenic mouse models, subchronic real-world PM2.5 exposure increases phosphorylated tau and hippocampal oxidative stress (measured as malondialdehyde) and causes cortical neuronal loss — even without frank cognitive impairment in the short term. Ferroptosis — iron-driven, ROS-mediated cell death dependent on the NRF2/Keap1/p62 axis — is an additional PM-relevant death mechanism in neurons.
In the lung, chronic PM2.5 exposure drives epithelial-mesenchymal transition (EMT) — converting epithelial cells into migratory, invasive, matrix-producing mesenchymal cells through coordinated activation of TGF-β/SMAD, PI3K/Akt, Wnt/β-catenin, NF-κB, ERK, JAK/STAT3, Notch, Hedgehog, HMGB1/RAGE, and aryl hydrocarbon receptor signaling — with PM2.5 components (heavy metal ions, PAHs, ROS) driving each of these oncogenic cascades and the cytoskeletal rearrangements that define malignant transformation. PM2.5 is thus both a promoter of pulmonary fibrosis and a lung carcinogen, acting through convergent molecular mechanisms that connect oxidative stress, inflammation, and epigenetic reprogramming to the hallmarks of cancer.