
1.5: Soil Chemistry
- Page ID
- 16609

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Darshani Kumaragamage, Jim Warren, and Graeme Spiers
Learning Objectives
On completion of this chapter, students will be able to:
- Define and describe the importance of the major soil chemical properties in sustaining life on Earth
- Describe and compare the structures, charge development, and properties of organic and inorganic soil colloids
- Explain the influence of soil minerals on soil properties
- Define cation exchange capacity, explain the importance of cation exchange and anion adsorption, and identify the factors influencing ion exchange in soils
- Discuss the causes of soil acidity and alkalinity, characteristics of strongly acidic and alkaline soils, and their effect on plant growth
- Describe the causes of soil salinity, major characteristics of saline, saline-sodic and sodic soils and reclamation of salt-affected soils
- Describe the importance of redox reactions in nutrient dynamics and greenhouse gas emissions
- Explain the basic principles in measuring cation exchange capacity, pH, electrical conductivity and redox potential in soils, and the interpretation of their values
INTRODUCTION
Soil chemistry is a branch of soil science that deals with the chemical composition, chemical reactions and chemical properties in soils. Abiotic phases of the soil include solids (organic matter and inorganic minerals), liquids (soil water), and gases (soil air), while the biotic phase consists of living organisms. Soil, therefore, is a dynamic living system in which ions and molecules constantly may move from one phase to the other, while interacting with each other.
Soil chemical properties are based on the concentrations, and/or proportions of dissolved species in soil water and/or on the ion exchange complex. Soil chemical properties such as cation exchange capacity (CEC), pH, redox potential (Eh or pe), and electrical conductivity (EC) are important as they influence nutrient availability, plant growth, fate of pollutants, biological activity, etc. For example, CEC is a measure of the amount of negative charge capable of adsorbing cations on mineral or organic surfaces, usually expressed in centimoles of charge [cmol(+)] per kg of soil. Soil pH is a measurement of the concentration of H+ in the soil solution, while the Eh (or pe) is related to concentrations of oxidized/reduced species of redox-sensitive elements such as iron (Fe3+/Fe2+) . Electrical conductivity (EC) is another important soil property that provides an indication of the abundance of soluble salts in a soil.
Most of the important chemical properties of soils are controlled by reactions that occur between the soil solution and surfaces of colloidal (particles <0.002 mm diameter; Brady and Weil, 2010) soil mineral particles and soil organic matter. In the first part of this chapter we will focus on the chemistry of the soil solution and the colloidal fraction. The remaining sections of the chapter will focus on important soil chemical properties; namely, CEC, anion exchange, soil pH, EC, and Eh, with emphasis on their importance in sustaining life on Earth.
SOIL Solution
Soil water, including dissolved solutes, is referred to as the soil solution and is the lifeblood of soil. All chemical reactions, mineral precipitation/dissolution reactions, ion exchange reactions, redox reactions, and nutrient uptake by plants occur in, or are mediated by the soil solution. Without soil water, very few chemical or biological reactions would occur in soil; in fact, there would be no life on this planet.
The soil solution is very complex and contains a large variety of cations and anions (as both as free ions and complex ions) as well as dissolved organic molecules, usually in low concentrations. For soil nutrients to be taken up by plant roots, they must exist in a dissolved form in the soil solution. At least 17 elements are required to support the growth of most plants (see Chapter 7), with an additional four elements essential for the growth of some plants (Havlin et al., 2013). For example, some important cations and anions that plants absorb from soil solution are: nitrogen as ammonium (NH4+) and nitrate (NO3–), phosphorous as several phosphate species (e.g., HPO42- and H2PO4–), potassium (K+), calcium (Ca2+), magnesium (Mg2+), and sulphur as sulphate (SO42-). All nutrients are absorbed from the soil solution by plant roots.
The soil solution also contains other non-nutritive ions and molecules, some of which are innocuous, while some are toxic, depending on concentration. These include some trace elements (e.g., lead, arsenic), pesticides (e.g., chlorpyrifos, glyphosate), antimicrobial ions and compounds (e.g., disinfectants, antibiotics), and complex organic molecules. These may also be taken up by plant roots. Nutrients and other chemicals in the soil solution may also be lost from the soil through surface runoff and/or downward leaching, particularly under excessive rainfall and irrigation events.
SOIL COLLOIDS
The soil colloidal fraction is defined as that fraction of the soil made up collectively of small (<0.002 mm) inorganic and organic particles (Brady and Weil, 2010). The high specific surface area and the presence of electric charges on most particles are responsible for important soil characteristics such as swelling capacity, the ability of the soil to retain water, cation and anion adsorption and ion exchange.
The importance of colloids in soil relates to the relationship of their size with the amount of surface area available to react with the soil solution. Table 5.1 shows the relationship between particle size and the magnitude of surface area associated with particle size range for a clay loam soil. Specific surface area is defined as the surface area per unit mass of the solid particle and expressed as m2 g-1 of soil. As can be seen in Table 5.1, even though this soil contains equal proportions of sand, silt, and clay by mass, by far the majority of the surface area is associated with the smallest size fraction.
Table 5.1. Relationship between particle size range and specific surface area of soil particles.
| Particle Size Fraction | Size Range | Median Diameter | Percent of Clay Loam Soil a |
Specific Surface Area b |
Percentage Surface Area |
|---|---|---|---|---|---|
| (µm) | (µm) | (%) | (cm2 g-1) | (%) | |
| Sand | 2000-50 | 1000 | 33.3 | 22.6 | 0.2 |
| Silt | 50-2 | 25 | 33.3 | 90.9 | 0.8 |
| Clay | <2 | 1 | 33.3 | 11,320 | 99 |
| a Assumed a clay loam soil with equal proportions of sand, silt and clay. b Calculated based on uniform spherical particles with median particle diameter. | |||||
Mineral phases in the colloidal or clay-size fraction are primarily phyllosilicate and oxy-hydroxide minerals (Figure 5.1). Organic colloids are also in the clay-size range and are composed of small particles of humus that are relatively stable and resistant to further biological decay. These diverse colloidal particles may either exist independently in the soil environment or they may occur in association in organo-mineral complexes (Newman and Hayes, 1990).
Phyllosilicates
It is important to understand that there are differences among aluminosilicate clay minerals (also called phyllosilicates or layer silicates) that make up the vast majority of mineral colloids and that these impart substantial differences in important soil properties, such as ion exchange.
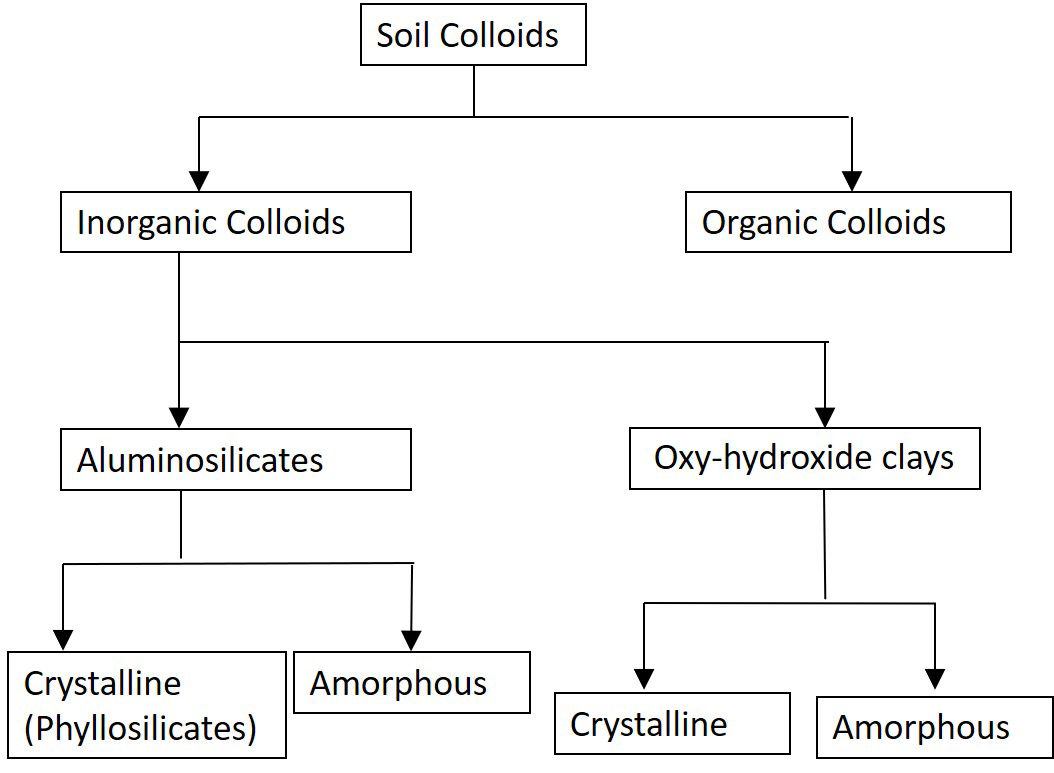
Phyllosilicates (phyllon meaning leaf or sheet in Greek) are crystalline minerals, with a structure consisting of layers of repeating atomic units. There are two fundamental units composed of layers of oxygen and hydroxyl groups bonded with silicon, aluminum, magnesium and other cations. The fundamental layers are composed of tetrahedral and octahedral sheets that occur in different combinations to form the different clay minerals.
Tetrahedral sheets are composed of repeating tetrahedron units, each typically containing a silicon atom surrounded by four oxygen atoms: one apical oxygen atom and three basal oxygen atoms forming a four-sided (tetrahedral) configuration (Figure 5.2A). Tetrahedral sheets extending in two dimensions are formed by sharing the basal oxygen atoms between silicon atoms (Figure 5.2B).
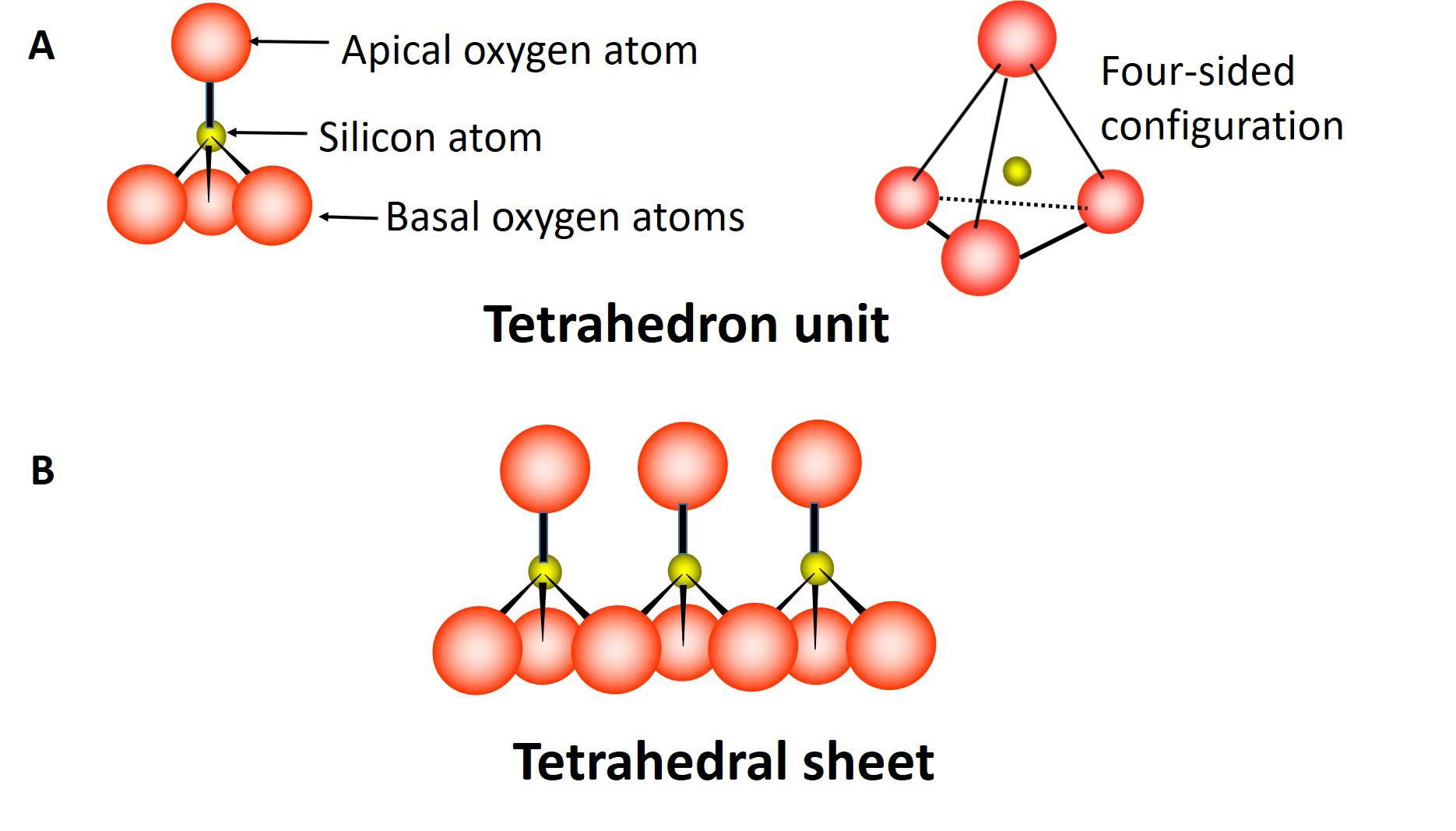
The octahedral sheet is composed of multiple octahedron units (Figure 5.3A), each unit containing aluminum (or magnesium) atoms surrounded by six oxygen atoms and/or hydroxyl groups. Three oxygen atoms (or hydroxyl groups) occur in the lower plane, the remaining three oxygen atoms (or hydroxyl groups) occur in the upper plane with the aluminum or magnesium atoms sandwiched in between, forming an eight-sided (octahedral) configuration. These units combine by sharing oxygen atoms to form an octahedral sheet extending in two dimensions (Figure 5.3B).
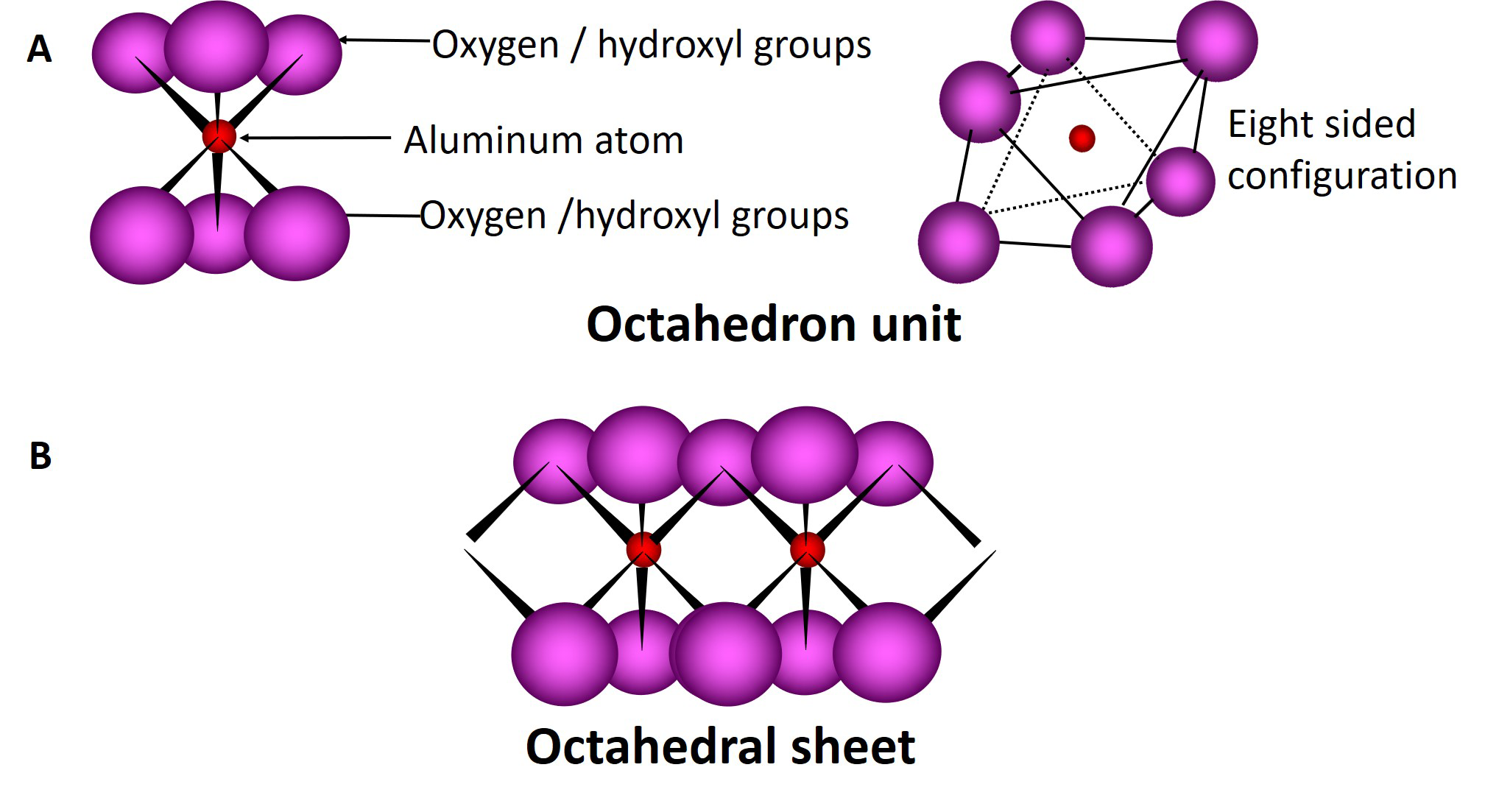
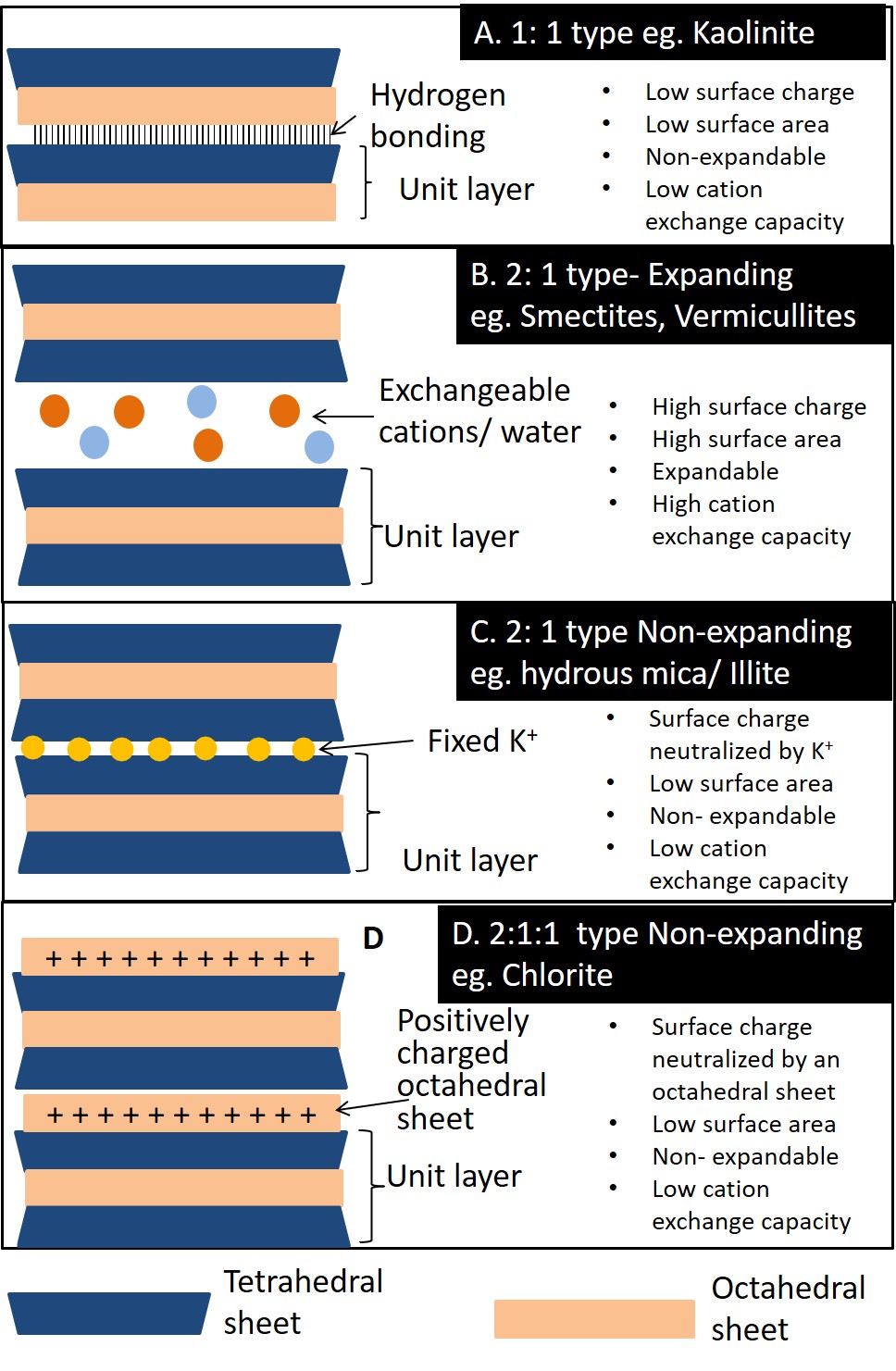
Tetrahedral and octahedral sheets combine to form unit layers of different phyllosilicates through the sharing of the apical oxygen atom of the tetrahedral sheet with oxygen atoms in the octahedral sheet. Phyllosilicates composed of one tetrahedral sheet and one octahedral sheet are called 1:1 type phyllosilicates; those composed of one octahedral sheet sandwiched between two tetrahedral sheets are called 2:1 type phyllosilicates (Figure 5.4). The ideal unit cell formula for a 1:1 clay mineral is Si4Al4O10(OH)8, while that of a 2:1 clay mineral is Si8Al4O20(OH)4.
Phyllosilicates consist of many layers (1:1 type or 2:1 type) stacked one on top of the other with different forms of bonding between the layers. The region located between two adjacent layers is called the interlayer region. The basic chemical behaviour and physical properties of the different phyllosilicates are dictated in large part by the characteristics associated with the interlayer region of the clays.
Isomorphous Substitution
In addition to their large surface area (see Table 5.1), the phyllosilicates are also electrically charged due to substitution of the central cations within the tetrahedral or octahedral sheets by other ions of similar size (see Table 5.2) but different valence. This imparts a net negative charge to the structure of the mineral.
Isomorphous substitution is defined as “replacement of one atom by another of similar size in a crystal structure without disrupting or changing the crystal structure of the mineral” (Canada Department of Agriculture, 1976). When a substituting cation is of a lesser valence than the cation it is replacing, there is an excess of negative charges associated with the oxygen atoms surrounding it and therefore a net negative charge is imparted to the structure. For example, trivalent aluminum (Al3+) may substitute for some of the tetravalent silicon (Si4+) in the tetrahedral sheet. The lesser charge of the aluminum compared to silicon creates a charge deficit and a negative charge within the tetrahedral sheet of the structure. Similarly, in the octahedral sheet, Mg2+, Fe2+, or other divalent cations, may substitute for some trivalent Al (Al3+), resulting in an extra negative charge associated with the octahedral sheet of the structure. These negative charges attract positively charged cations to the surface of the minerals to neutralize the negative charge. These charge neutralizing cations are located mostly within the interlayer region of the phyllosilicates. The abundance of negative charge sites (i.e., the charge density), the location of the negative charges (tetrahedral vs. octahedral sheet), and the type of cation in the interlayer region neutralizing the charge (e.g., Na+, Ca2+, Mg2+, K+ or other) determines the degree to which the phyllosilicates expand or swell, and their capacity to retain cations.
Table 5.2. Radius of common cations and location in phyllosilicate structures
| Cation | Radius (nm) | Location |
|---|---|---|
| Si4+ | 0.041 | tetrahedra |
| Al3+ | 0.05 | tetrahedra, octahedra, interlayer |
| Fe3+ | 0.064 | octahedra |
| Fe2+ | 0.076 | octahedra, interlayer |
| Mg2+ | 0.065 | octahedra, interlayer |
| Ca2+ | 0.099 | Interlayer |
| Na+ | 0.095 | Interlayer |
| K+ | 0.133 | Interlayer |
Phyllosilicate Minerals
There are about 50 species of phyllosilicate minerals, but only five are common in Canadian soils (see Chapter 14). These are sketched in Figure 5.5 and described as follows:
Kaolinite (Figure 5.5A) is a 1:1 type clay mineral held together by hydrogen bonds in the interlayer region. Hydrogen bonding occurs between the hydroxyl in the octahedral sheet of one unit layer, and the (basal) oxygens in the tetrahedral sheet of the adjacent layer. The hydrogen bonds hold the layers closely together to form a very stable structure. This combined with very little isomorphous substitution inhibits the intrusion of cations and water into the interlayer region. This mineral is non-expandable since water molecules cannot enter the interlayer space. Kaolinite exhibits very little swelling when wet, or shrinkage when dry. The interlayer is not exposed and therefore the specific surface area is low. With almost no isomorphous substitution, kaolinite minerals have a low surface charge and low cation exchange capacity.
Can You Dig It!
Rocks that are rich in kaolinite are known as kaolin which is derived from the Chinese word “Gaoling” (literally meaning ‘High Ridge’), which is the name of a village in southeastern China where kaolinite was mined historically. Kaolinite in its pure form is white and its minimal shrink/swell characteristic and ability to not crack when dried made it highly prized for making porcelain and “fine China” dishware.
Smectite (Figure 5.5B) is a group of expandable 2:1 type phyllosilicates. Montmorillonite and beidellite are the two most common species of the smectite group of minerals in Canadian soils. The amount of isomorphous substitution in both minerals is moderate compared to other 2:1 type phyllosilicates (e.g. hydrous mica), and the two species are differentiated based on the location of the layer charge in the structure. Most of the layer charge in montmorillonite is a result of isomorphous substitution in the octahedral sheet, whereas in beidellite the layer charge derives mostly from isomorphous substitution in the tetrahedral sheet. Hydrogen bonding does not occur in 2:1 clay minerals since there are no hydroxyl groups associated with the basal oxygens of the tetrahedral sheets of adjacent layers facing each other. Thus, the structural layers are only held together by electrostatic forces of whatever neutralizing cation happens to be in the interlayer region to neutralize the negative charges associated with isomorphous substation within the layer structures. Exchangeable cations in the interlayer are usually surrounded by six water molecules in octahedral coordination. Smectite minerals have a high cation exchange capacity. Since the layers are loosely bound, both water molecules and cations can freely enter the interlayer space. Therefore, these clays have a high specific area, and are expandable; swelling when wet and shrinking when dry.
Vermicullite (Figure 5.5B) is a 2:1 type phyllosilicate similar to smectite but with a layer charge intermediate between hydrous mica/illite (see below) and the smectites. The mineral has limited expandability, with moderate shrink/swell characteristics. Most isomorphous substitution occurs in the tetrahedral sheet imparting a high surface charge and a high cation exchange capacity. The structure is capable of trapping K+ ions in the interlayer region similar to hydrous mica/illite, but as the layer charge is slightly lower, the trapped K+ can be released back into the soil solution unlike hydrous mica/illite.
Hydrous Mica also called illite (Figure 5.5C) is another 2:1 type clay mineral, Hydrous mica is derived from the partial weathering of mica, a primary mineral common in the granitic rocks of the Canadian Shield. The structure of hydrous mica/illite is similar to the smectites and vermicullite, except that the layer charge is higher with isomorphous substitution occurring almost exclusively in the tetrahedral sheet where Al3+ is substituted for Si4+. Potassium almost exclusively occupies the interlayer region, neutralizing the layer charge and holding the structure together. The potassium ion (K+) is almost an exact fit for the hexagonal shaped holes that occur in the tetrahedral sheets of the 2:1 layers, and thus gets even closer to the charged sites and strongly binds adjacent layers together. This prevents entry of water into the interlayer region and renders minerals of the mica group non-expandable. So even though the layer charge is higher than the smectites and vermiculite, the cation exchange capacity of hydrous mica/illite is low, since the charge is neutralized by K+.
Chlorite (Figure 5.5D) is a 2:1:1 (or 2:2) type phyllosilicate. The mineral has a 2:1 type structure, but with an additional octahedral sheet—composed of an aluminum, magnesium or iron hydroxide layer in six-fold (octahedral) coordination—in the interlayer region between the 2:1 type layers. This interlayer hydroxyl layer does not share oxygens with the main 2:1 layer, instead it is hydrogen bonded to the 2:1 layer; consequently, the mineral is non-expandable. The amount of isomorphous substitution in the structure is variable, but the charges are neutralized by the positively charged octahedral sheet in the interlayer, thus the ion exchange capacity is low.
Amorphous aluminosilicates are yet other aluminosilicates found in soil but these are not phyllosilicates. These are described as amorphous or as minerals of short-range order, meaning they lack the defined structural units of the phyllosilicates. Allophane and imogolite are examples of amorphous aluminosilicate minerals. These minerals are formed in abundance in volcanic ash soils found in countries such as Japan and New Zealand, but they are also being found in very small amounts in some Podzolic and Solonetzic soils in Canada. Amorphous aluminosilicates are non-expandable. They are extremely small (<4 nm diameter) with a very high specific surface area and a very high ion exchange capacity.
Oxy-hydroxide Minerals
Oxy-hydroxide minerals (sesquioxides) are mainly oxides, hydroxides and hydrous oxides of iron, aluminum, and manganese, and are among the youngest mineral phases found in soil. Some are well crystallized (e.g., gibbsite, hematite, goethite), while others are amorphous (e.g., ferrihydrite). Crystalline oxide clays are composed of oxygen and/or hydroxyl groups, usually in an octahedral arrangement with iron, aluminum, or manganese. The octahedral structures of these minerals are held together by H-bonding. Non-crystalline (amorphous) oxy-hydroxides are complex iron, aluminum, and manganese minerals that are typically precipitated very quickly from the soil solution in response to wetting and drying cycles. Precipitation of these minerals occurs when the solubility product for a combination of cation and anion concentrations is exceeded, and usually other solutes including carbonate, phosphate and sulphate may be trapped in the structure during the rapid precipitation. Amorphous oxy-hydroxides are metastable and convert slowly to their more crystalline forms.
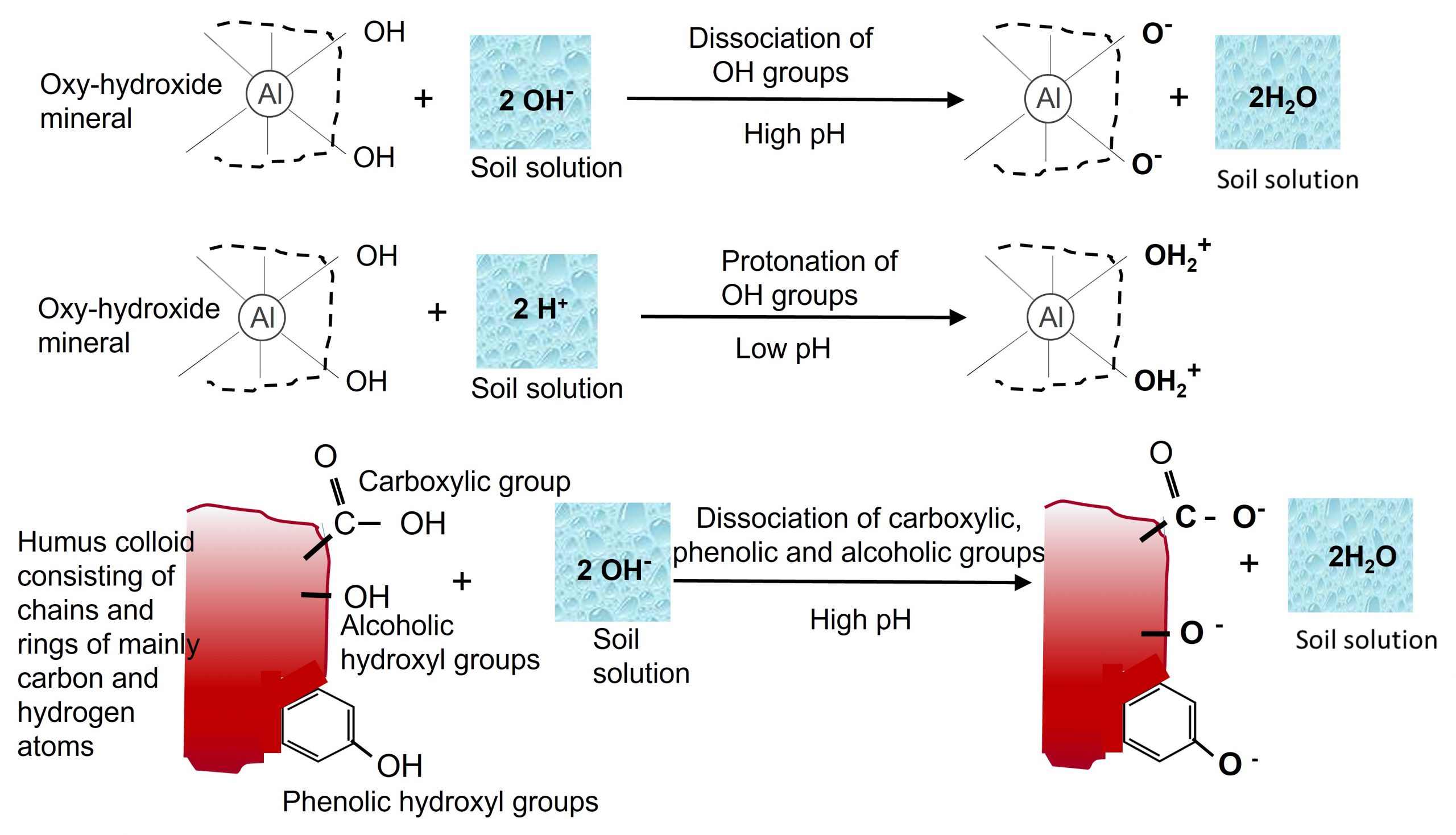
Both crystalline or amorphous oxy-hydroxide minerals do not carry permanent negative charges but do have a high surface charge associated with surface hydroxyl groups. As such, the exchange capacity of oxy-hydroxides is very pH dependent with the variation due to protonation of hydroxyl groups at low pH producing positive charges, and dissociation of hydroxyl groups at high pH producing negative charges. Oxy-hydroxide minerals are amphoteric due to their variable cation exchange capacity; meaning they typically have a net cation exchange capacity at high pH and a net anion exchange capacity at low pH.
Oxy-hydroxides are usually found in small quantities in young Canadian soils, but are more common in highly weathered soils found in warm, humid regions in other parts of the world. They are highly reactive and have a very high specific surface area. Presence of these minerals significantly influences soil properties, such as ion adsorption and exchange.
Organic Colloids (Humus)
Organic colloids, generally known as humus, consist of highly decomposed organic matter. These colloids have a complex structure, and are chemically composed mainly of carbon, hydrogen and oxygen. On the molecular scale, humus may be described as a complex polymeric mixture of random organic components and is probably one of the least understood aspects of soil chemistry. Organic colloids may be highly reactive chemically, with high water holding capacities allowing absorption of water. Compared to inorganic colloids, organic colloids have greater specific surface area and higher surface charge. Surface charges are predominantly negative and pH dependent arising from partial dissociation of hydroxyl (-OH), carboxyl (-COOH), and phenolic groups, which are associated with central carbon structures of varying size and complexity. On a per unit mass basis, organic colloids impart a greater influence on soil properties than inorganic colloids even though their abundance in soil is generally far less than that of the inorganic colloid fraction.
Charges on Soil Colloids
Charges on soil colloids arise from the following two mechanisms:
- Isomorphous substitution within the crystalline structure of phyllosilicates – leading to formation of mostly negative, permanent charges. As described earlier in this chapter, isomorphous substitution is the replacement of one atom by another of similar size in a crystal lattice without disrupting or changing the crystal structure of the mineral. The replacement of a higher valent cation with a lower valent cation (e.g., substitution of Mg2+ for Al3+ in the octahedral sheet) results in an unbalanced negative charge on the mineral surfaces. These negative charges are permanent and thus are not affected by pH.
- Protonation/Deprotonation of functional groups on the surfaces of oxy-hydroxide clays, the edges of phyllosilicate clay minerals and organic compounds, give rise to positive and negative charges, referred to as pH dependent (variable) charge. Protonation of functional groups occurs under acidic (low pH) conditions whereas dissociation of hydroxyl protons predominates under basic (high pH) conditions (Figure 5.6). Thus, the relative amounts of positive and negative surface charge vary depending on soil pH, with negative charges predominating under high pH conditions and positive charges predominating at low pH conditions. The pH at which the number of positive charges is equal to the number of negative charges is referred to as the point of net zero proton charge or zero point of charge. Humus particle surfaces are similar to inorganic colloids, containing functional groups (e.g., carboxyl [-COOH], phenolic [R-OH]) that can dissociate (deprotonate) depending on the pH, giving rise to a negative charge on organic colloids. As the pH increases, dissociation is greater, and the negative charges increase. In acidic soils, conditions favour the non-dissociated functional group, thus negative charges are low.
Influence of Soil Colloids on Soil Properties
The amount and type of inorganic and organic colloids present in the soil has a profound influence on soil properties. For example, soils containing a high proportion of humus have high water holding capacities. Soils having a high proportion of freely expanding clay minerals, such as smectite, shrink when dry and swell when wet. Vertisols, fine textured soils containing >60% clay of which at least half is smectite, are typically very sticky when wet and form wide cracks when dry.
Phyllosilicates, oxy-hydroxides, and humus particles have electrical charges on their surfaces, which contribute to the total charge in the soils, and thus to the cation exchange capacity. The ranges of CEC associated with the different soil colloids are presented in Table 5.3. For example, a soil containing 40% smectite clay with CEC of 100 cmol(+) kg-1 may contribute up to 40 cmol(+) kg-1 to the total CEC of the soil. Given its high charge density, humus can be a significant contributor to the total CEC even in mineral soils—particularly under neutral to high pH conditions where pH dependent negative charges dominate. Depending on the characteristics of the humus, the pH, and interactions of humus with other soil particles, a 3% humus content (assuming a CEC of 400 cmol(+) kg-1), may contribute up to 12 cmol(+) kg-1 to the CEC of the soil. The contribution of iron oxy-hydroxide clays to CEC is also pH dependent. However, compared to humus and phyllosilicates with high CEC, the contribution of iron oxy-hydroxides towards soil CEC is negligible. Conversely, oxy-hydroxides together with 1:1 type layer silicates may exhibit positive charges under acidic conditions and are thus a main source of anion exchange capacity.
Table 5.3. Ranges of CEC in different soil colloids. Adapted from Brady and Weil (2010) and Soil Survey Staff (2014)
| Colloid | CEC Range [cmol(+) kg-1] |
|---|---|
| Kaolinite | 2 to 16 |
| Chlorite | 10 to 40 |
| Hydrous Mica/Illite | 20 to 40 |
| Montmorillonite | 60 to 100 |
| Vermiculite | 100 to 150 |
| Organic matter (humus) | 150 to 400 |
| Amorphous aluminosilicates | 5 to 350 |
| Fe/Al oxy-hydroxides | ~0 to 3 |
CATION EXCHANGE IN SOILS
Negatively charged surfaces on soil colloids attract cations by electrostatic forces. Cations retained by soil colloids may be eventually released to the soil solution through exchange with other cations in the soil solution. The cations most commonly held by soil colloidal particles are Ca2+, Mg2+, K+, Na+, H+ and Al3+. Of these, Ca2+, Mg2+, K+, and Na+ are so-called base-forming cations, while H+ and Al3+ are acid-forming cations. Exchangeable H+ and Al3+ are found in increasing abundance in acidic soils.
Cation exchange is a process by which cations in the soil solution exchange with cations attached to the surfaces of clay minerals and humus. Cation exchange occurs on a charge-for-charge basis as illustrated in Figure 5.7.
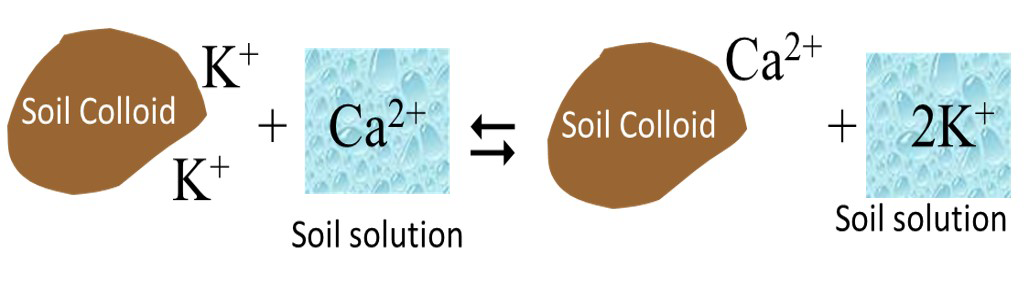
Cation exchange reactions are stoichiometric. Thus, for divalent-monovalent exchanges (e.g., exchange between Ca2+ and K+) one divalent cation exchanges with two monovalent cations. The reaction is rapid (almost instantaneous) and reaches equilibrium quickly. The cation exchange reaction is usually reversible, unless the exchanged cation is fixed in the interlayer space of a phyllosilicate (e.g., K+ in hydrous mica/illite).
Cation exchange capacity (CEC) is defined as the sum of total exchangeable cations that a soil can adsorb (Brady & Weil, 2010). The CEC is usually expressed in centimoles of positive charge per kilogram of exchanger (cmol(+) kg-1) or millimoles of positive charge per kilogram of exchanger (mmol(+) kg-1). Thus a soil with a CEC of 10 cmol(+) kg-1 can adsorb 10 cmol of a monovalent cation (e.g., Na+) or 5 cmol of a divalent cation (e.g., Ca2+). In this example, if Na+ exchanges with another monovalent cation (e.g., K+), 10 cmol of K+ would exchange with 10 cmol of Na+. However, if the exchange is with a divalent cation (e.g., Ca2+) only 5 cmol of Ca2+ will exchange with 10 cmol of Na+.
Since it is the negative charges (including pH dependent negative charges) in the soil colloids that are responsible for the CEC of a soil, the amounts of inorganic and organic colloids, the type of clay minerals present, and the soil pH are the primary determining factors controlling the CEC of a soil.
Can You Dig It!
Ion exchange in soils is important to sustain life on Earth. Because of ion exchange, plant nutrients are retained in soil and released when needed by plants. Thus ion exchange occupies a central position unique to soils in their ability to supply plant nutrients and act as a filter to retain contaminants from reaching groundwater. Ion exchange retains toxic compounds, reducing their mobility, thus, protecting water resources from pollution.
Determination of Cation Exchange Capacity
The CEC of soil is generally estimated by saturating all cation exchange sites with an index cation and then replacing and measuring the amount of the index cation released to solution. The first step is to saturate a known mass of oven-dried soil with an index cation (e.g., Na+ or NH4+). This is accomplished by equilibrating the sample with a solution containing the index cation buffered at pH 7.0 (usually sodium acetate or ammonium acetate). After ensuring that all cations are displaced by the index cation, the excess index cation is removed from the soil sample by leaching with a non-polar solution such as 95% ethanol. The amount of index cation held by the exchange sites in the sample must then be determined by leaching the soil with a solution containing a different cation (e.g., K+) to replace the index cation on the exchange sites. Then the concentration of index cation in the leachate is measured and used to calculate the CEC of the soil (Hendershot et al., 1993).
Base Saturation
The proportion of CEC saturated by base-forming cations (typically Ca2+, Mg2+, K+, and Na+) is known as base saturation. Base saturation percentage is calculated using the equation:
(1) 
Both the CEC and the sum of exchangeable base-forming cations should be expressed in the same units (e.g., cmol(+) kg-1). Since the dominant exchangeable base-forming cations in soils are Ca2+, Mg2+, K+, and Na+, the equation is usually written as:
(2) 
Note that for divalent cations such as Ca2+ and Mg2+, exchangeable cations in cmol (+) should be calculated considering the charge; i.e, 1 cmol Ca2+ = 2 cmol of charge (+). Base saturation is positively related to soil pH. If the pH is low (<7) then the base saturation is also low. And, as soil pH increases, base saturation also increases approximately linearly between pH 4 and 7.
ANION ADSORPTION AND EXCHANGE
Soils are able to retain anions in a similar fashion to retaining cations, particularly in the lower pH range when the surfaces of some colloids (e.g., oxy-hydroxides) have positive charges. Retention of anions through adsorption varies from one anion to another. Some anions are retained through non-specific adsorption mechanisms, while others are retained through specific adsorption mechanisms.
Non-specific adsorption refers to an anion that is retained by positively charged sites and the anions are weakly held through electrostatic forces. This type of adsorption is common for anions like chloride (Cl–) and NO3–. These anions are weakly held on positively charged surfaces and they can freely exchange with other anions in soil solution; consequently, they may be easily lost through leaching. For non-specific adsorption to occur, the colloidal surfaces must be positively charged; thus, soil containing minerals with pH dependent charges have a high anion exchange capacity, particularly at low pH. Soils of the tropics contain high proportions of oxy-hydroxides and kaolinite, which exhibit positive charges under low pH conditions, thus in these soils, anion adsorption through non-specific mechanisms can be high.
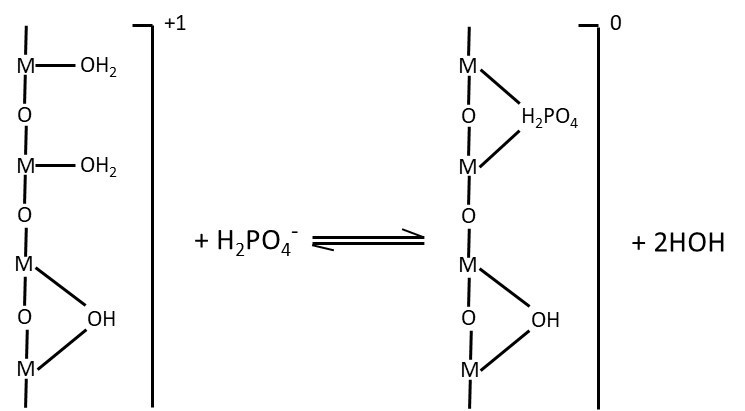
Specific adsorption occurs when the ion becomes chemically bonded to the surface of the colloid. Mechanisms are specific to the anion being adsorbed. For example, phosphate anions (e.g., H2PO4–) may be specifically adsorbed to certain colloidal surfaces through exchange of a surface hydroxyl on the colloid for an oxygen atom that is part of the anion (Figure 5.8). The strongly bonded anion is not mobile in soils and is less available to plants. Specific adsorption is only minimally reversible, resulting in permanent loss of the ion from the soil solution and from uptake by plants. In extreme cases, the original mineral surface may become completely coated with the adsorbed ion. The common anions that are retained in soil through specific adsorption are phosphate, sulphate, silicate, and carbonate.
SOIL pH
Soil pH is a measure of the activity (cf. concentration) of hydrogen ions in the soil solution and describes the acidity or alkalinity in the soil. For our discussion herein we define activity as “effective” concentration, whereby activity is roughly equal to concentration in dilute solutions but generally deviating from concentration (typically becoming less than the concentration) as the total concentration of salts dissolved in the soil solution increases. For current purposes, we assume that activity and concentration are equal.
The standard pH scale spans from 0 to 14, with a pH value of 7 being neutral. Pure water has a pH value of 7.0. Values of pH less than 7 are acidic and become increasingly acidic as values approach zero. Values of pH greater than 7 are alkaline, or basic, and become increasingly alkaline as values approach 14. The pH scale is logarithmic, which means that for every one-unit change in pH there is a 10-fold change in the activity of hydrogen ions in the soil solution. For example, a soil with pH 6 has ten times more hydrogen ions in solution than a soil at pH 7, and a soil with pH 8 has ten times less hydrogen ions than a soil at pH 7. A comparison of soil pH values for some soils with some common acids and bases are shown in the Figure 5.9.
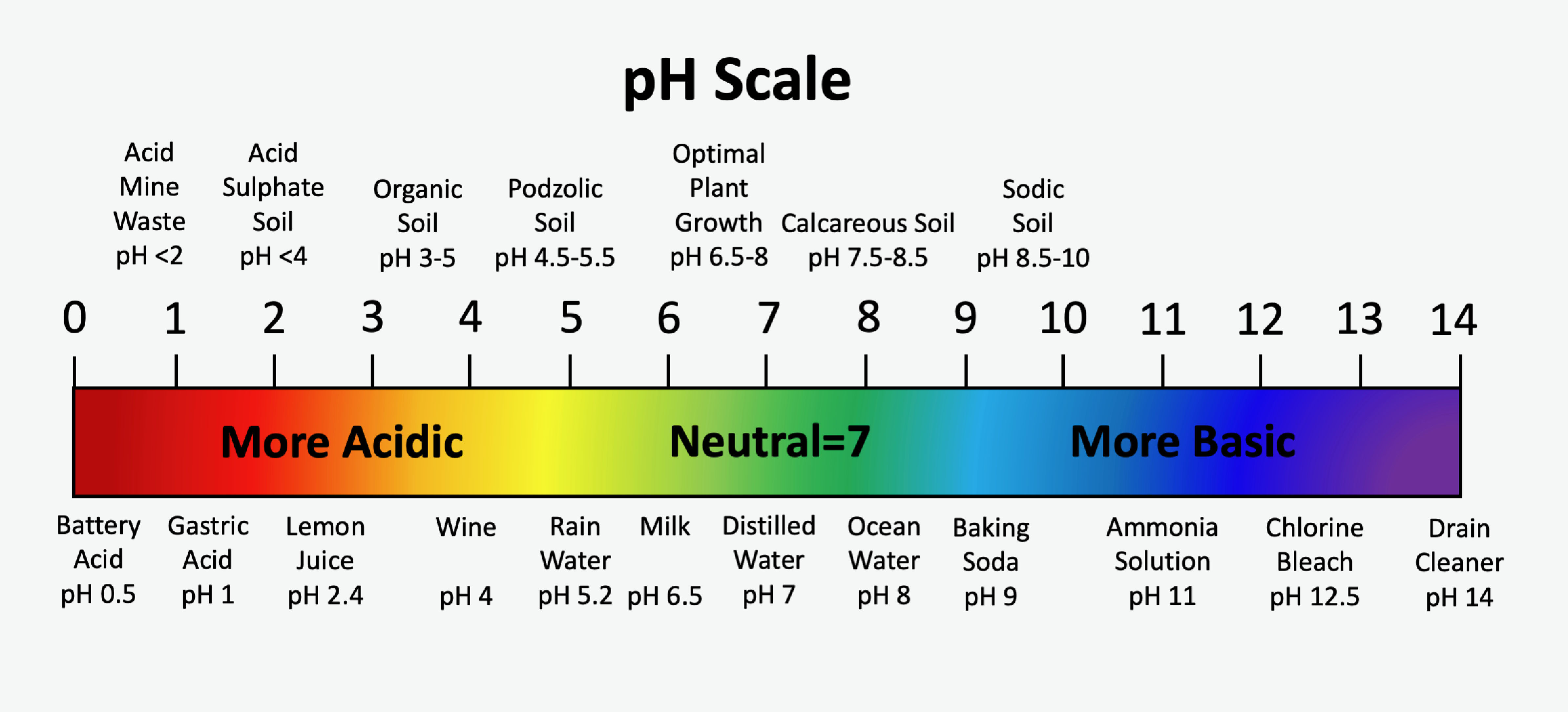
The primary factors influencing the natural soil pH are the geological parent materials of the soil and reaction with carbon dioxide (CO2) dissolved in meteoric water. Organic acids derived from the decay of vegetation also influence soil pH. Generally, mineral soils in Canada are formed on one of two very broadly defined parent materials: those developed on granite-based parent materials, commonly referred to as acidic parent materials, and those developed on carbonate-based parent materials, commonly referred to as alkaline parent materials. Granitic-based parent materials are derived mostly from rocks of the Precambrian Shield, while carbonate-based parent materials, typically limestone and dolostone (dolomite), are derived from Paleozoic aged rocks or younger. Soils formed from granitic-based parent materials are generally in the acidic pH range. Conversely, soils formed on carbonate-based parent materials typically have soil pH values in the neutral to alkaline range.
Rainwater equilibrates with atmospheric gases, particularly CO2, and has a pH of approximately 5.6. Other factors such as atmospheric pollutants (e.g. sulphur dioxide which hydrolyzes to sulphuric acid) typically drive the pH lower. The general trend for all soils is to be driven to the acidic end of the pH scale with increasing length of exposure to precipitation, which may cause leaching of basic cations. Granitic parent materials generally contain very little basic mineral material and therefore the pH of granitic-based soil is easily driven to the acid end of the pH scale. Podzolic soils, typically developed on granitic parent materials, have pH values in the range of 4.5 to 6.0. Although it may appear somewhat counter intuitive, the pH of soils containing carbonate minerals are buffered against the acidifying effects of dissolved CO2 in the soil solution. Calcareous soil materials typically have pH values in the range of 7.5 to 8.5.
Soil pH may be buffered in some soils. That is, although pH reflects the concentration of H+ in solution at the time of measurement, there are additional sources of H+ on exchange sites referred to as “exchangeable acidity” that buffer and maintain the pH in solution. Through exchange of H+ between the soil solution and exchange sites, high CEC soils can buffer soil pH, which means they will resist a change in pH when an acidic or basic compound (e.g., a fertilizer) is added. Acidity, measured immediately in the soil solution, is referred to as “active acidity”, while acidity on the exchange sites is referred to as “reserve acidity”. Some soils are highly pH buffered with a large amount of reserve acidity (e.g., soils with high clay and/or organic matter content), while others are not (e.g., sandy soils with low organic matter content).
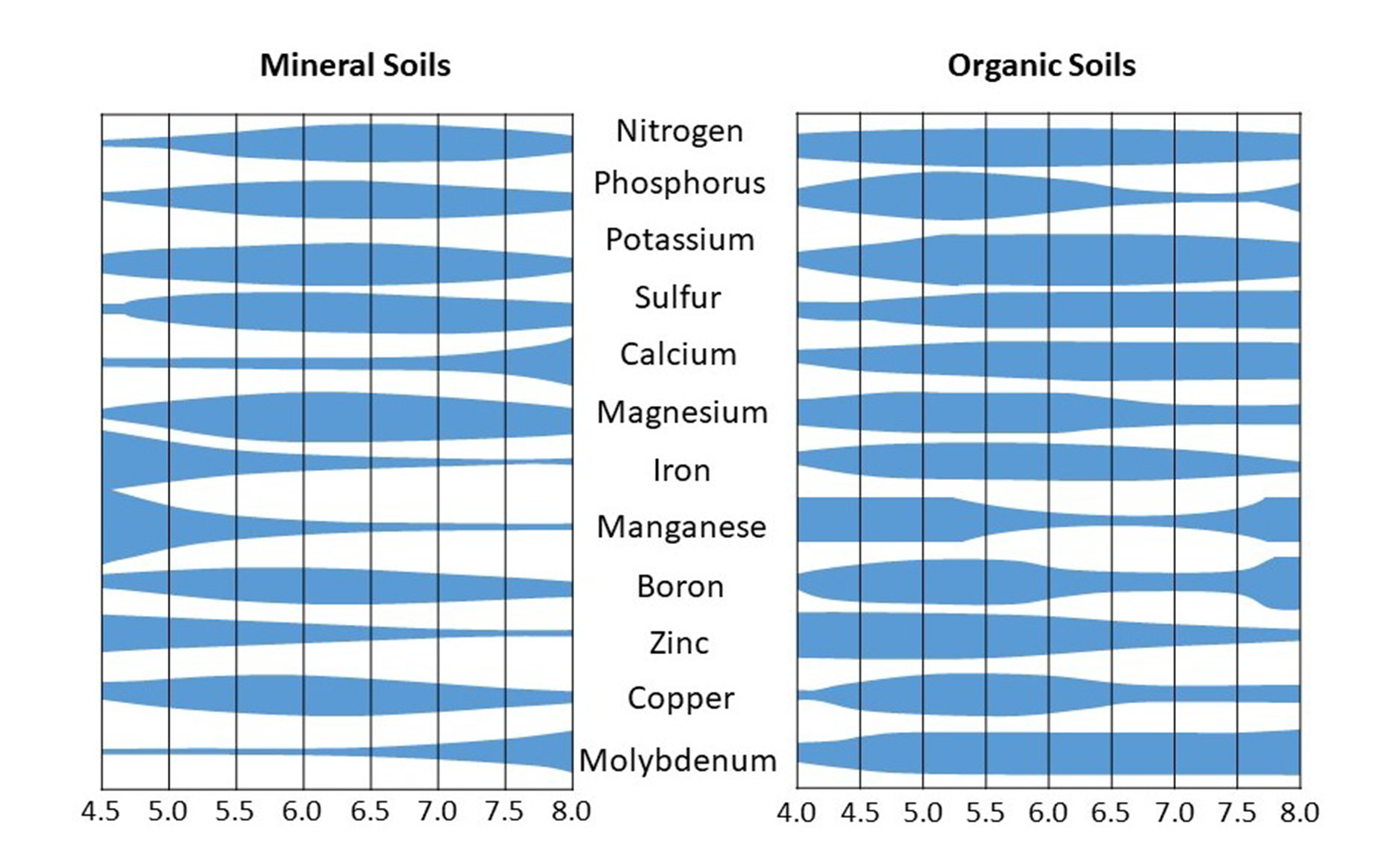
Soil pH affects plant nutrient availability. However, not all nutrients are available at a specific pH level. Soil pH values in the range of 6.5-8.0 are considered optimum for the availability of most nutrients to plants. Generally, extreme pH values are detrimental to nutrient uptake and plant growth. For example, acidic pH values increase the solubility of aluminum, manganese and iron, which are potentially toxic to plant roots when in high concentrations in the soil solution. These elements also may react with plant nutrients to render them unavailable to plants. Similarly, alkaline pH values result in excess amounts of calcium in the soil that also react with nutrients (e.g., phosphate) so they are unavailable for plant use. General ranges of nutrient availably with pH in mineral and organic soils are shown in Figure 5.10.
Acids and Bases
Soil pH values reflect the various reactions involving acid-base pairs in water, which itself dissociates:
H2O ↔ H+ + OH–
An aqueous solution is said to be neutral (pH = 7) when the concentration of protons [H+] exactly equals the concentration of hydroxyl ions ([OH–]), with both equal to 10-7 moles per litre.
An acid is any chemical substance that can donate a proton when dissolved in water, while a base is any substance that can accept a proton. When an acid loses a proton it forms the conjugate base:
HL ↔ H+ + L–
In the above expression HL is an acid capable of donating a proton (H+) to solution and L– is the conjugate base, capable of accepting a proton. In this expression, the base (L–) or conjugate base is also referred to as a ligand. The strength of an acid is the measure of its ability to dissociate in water and release protons to solution. A strong acid (e.g., sulphuric acid) is a substance that completely dissociates into protons and ligands.
A base is any chemical substance that can generate hydroxide ions (OH–) or takes up H+ ions in solution forming its conjugate acid.
B + H2O ↔ OH– + BH+
Humified soil organic matter, although perhaps still poorly understood in terms of specific structural/chemical makeup, is the source of surface functional groups that influence the chemistry of the soil. Some of the most important organic functional groups include carboxylic (–COOH), phenolic-OH, alcoholic-OH, carbonyl (C=O) and quinone groups, as well as amine (-NH2) and thiol (-SH) groups. The carboxylic acid, phenolic-OH and alcoholic–OH functional groups contribute to acidity and ion-exchange. These groups behave as weak acids in soil acting as a source of reserve acidity and serve as cation exchange sites. Other functional groups, such as aromatic acids, dissociate and function in a similar fashion.
Soil pH controls many of the chemical processes that take place in the soil, especially plant nutrient availability. Most soils have pH in the range of 5 to 8 with the optimal pH range for most agronomically important plants between 5.5 and 7.5, though many plant species have adapted to pH values outside of this range. For example, blueberries prefer soil with pH in the range of 4.3 to 5.5, growing very poorly in soils with pH in the alkaline range (pH>7).
If soil contains significantly more Na+ in the soil solution compared to Ca2+ or Mg2+, there may be a risk of sodium buildup. This ion buildup is of greater concern in arid or semi-arid regions (e.g., Canadian prairies) than in more humid regions (e.g., eastern Canada). Soils with naturally high sodium levels, or those that have received large quantities of sodium bicarbonate (Na2CO3) through irrigation, may have pH levels of 8.5 or higher. This high pH value is due to the presence of Na2CO3 precipitates in the soil which produce the elevated pH. This process is a characteristic of Solonetzic soils discussed later in this chapter.
Measurement of Soil pH
The pH of a soil is likely the most frequently measured soil chemical property. Soil pH measurements are typically made using a commercial pH meter equipped with a combination glass pH electrode. Combination electrodes are single electrodes that include both a glass electrode and a reference electrode. There are numerous methods for soil pH measurement, which depend on the application. Methods vary for the soil to solution ratios used; addition of an electrolyte and sample resting time prior to measurement. The methods include, but are not limited to measurements in: saturated paste; 1:1, 1:2, 1:2.5, 1:5, and 1:10 soil:water suspensions; 1:2 and 1:5 soil:0.01M CaCl2 suspensions; 1M KCl; and 1.0M NaF (Soil Survey Staff, 2014; Hendershot et al., 2008). The soil pH measurement is that of the solution in contact with the soil solids and not of the soil solids themselves (Hendershot et al., 2008). Soil pH values measured in electrolyte (e.g., 1:2 soil to 0.01 M CaCl2) suspensions are usually more stable and reproducible than those taken in water because the concentration of the electrolyte is constant. However, the addition of an electrolyte generally produces a decrease in soil pH compared to water; e.g., values measured in soil:water suspensions are typically 0.5–0.6 units higher than those in 0.01M CaCl2, and 0.7 units higher than those in 1M KCl. This is because of the inclusion of exchangeable (reserve) acidity introduced to solution through ion exchange reactions involving Ca2+ or other introduced ion with Al3+ and/or H+ ions on the exchange sites.
Although the pH of a soil containing a carbonate mineral should ideally be 8.2, soil pH measurements for these soils are commonly in the 7.6 to 8.5 range. This measured pH is a result of several factors. First, in addition to exchangeable ions such as Ca2+, Mg2+, Na+ and K+, the ion exchange sites on soil also contain small amounts of exchangeable aluminum and iron, which on entering the soil solution hydrolyze and react to reduce the measured soil pH. Second, although soils may contain carbonates, the mineral dolomite [Ca,Mg(CO3)2] reacts (dissolves) much slower compared to calcite (CaCO3), and the pH may not stabilize within the timespan (e.g. within an hour) of the measurement. Third, typical procedures for pH measurements (e.g., Hendershot et al., 2008) recommend allowing prepared sample suspension to stand for one hour prior to pH measurement. This procedure provides a standard reaction time allowing easy adaptation for routine high sample throughput operations in commercial soil laboratories that follow standard ISO17025 accredited analytical methods. Although this time frame is sufficient to allow for ion exchange equilibration, it may or may not be sufficient to allow full equilibration for dissolution of carbonate and equilibration with CO2 in the atmosphere. Other chemical reactions may also have an influence on the measured pH.
Soil pH Adjustment
The natural tendency is for soil to become more acidic through the natural acidity of rainwater, decay of organic matter and through application of fertilizers such as urea and anhydrous ammonia. However, soil pH may also be increased through additions of agricultural limestone. The timing and amount of agricultural “lime” applied depends on the crop to be grown, with lime application being commonly advised when soil pH values decrease below 6.0 (Munro, 2018). Finely ground limestone (primarily comprised of the mineral calcite; CaCO3) or dolostone (primarily comprised of the mineral dolomite; Ca,Mg(CO3)2) are the most frequently used liming products, though many other products with acid neutralizing capacity may be used.
Can You Dig It!

The Sudbury Regreening Program
A century of sulphur dioxide fumigation and nickel particulate deposition by base-metal smelters, coupled with fire, soil erosion, and enhanced frost action left thousands of hectares of barren land with eroded soils and stunted, open birch-poplar woodlands in the Sudbury area.
The relict soils of the area are coarse textured, eroded Podzols that are acidic, metal-rich, nutrient poor and drought-prone—all of which have contributed to limit plant recolonization. The drought vulnerability of seedlings germinating in the soils was further enhanced by aluminium toxicity limiting root growth, and further exacerbated by enhanced frost action in the medium textured soil that lacked the protection of surface humus forms previously lost as a result of wind and water erosion.
A revegetation program that detoxified the soils enough to allow for successful colonization was developed based on field research in the 1970’s. This research demonstrated that the surface application of ground dolomitic limestone [primarily CaMg(CO3)2] at 10 tonnes per hectare adjusted the soil pH from less than 3.0 to between 5.2 and 5.6. Application of the liming treatment, with or without an accompanying fertilizer and/or a grass-legume seed application, led to the natural colonization by woody species including birch, aspen, and willows. This colonization has further been aided by planting a mixture of native coniferous and deciduous species together in groups to form a “seed bank” for future colonization – with nearly 10 million trees being planted in the Sudbury region between 1976 and 2020.
Adjustment of soil pH towards the acid end of the scale may be required when growing certain crops such as potatoes, blueberries, azaleas or some other horticultural crops. Downward pH adjustment is typically accomplished with the addition of elemental sulphur, sulphur containing compounds or ammonia containing fertilizers; however, application of amendments to intentionally decrease soil pH is very uncommon.
SOIL SALINITY AND SODICITY
Soil salinity levels are controlled by the concentration of total dissolved mineral salts present in the soil solution. Salts occur naturally in both soil and water and may, under certain conditions, accumulate in soil. The accumulation of soluble salts in the soil is known as salinization. Dissolved salts that contribute to soil salinity include the cations Na+, Ca2+, Mg2+, and K+, and the anions Cl−, SO42−, HCO3−, CO32−, and NO3−.
Sodicity is related to salinity, but refers specifically to the concentration of Na+ in the soil solution relative to concentrations of Ca2+ and Mg2+. The degree of sodicity, estimated by calculation of the sodium adsorption ratio (SAR), is a measure of the concentration of Na+ in saturated soil extract relative to Ca2+ and Mg2+ concentrations. The SAR is calculated as the ratio of the Na+ concentration divided by the square root of average [Ca2+ + Mg2+] concentrations. The square root of [Ca2+ + Mg2+] concentrations is used since these are divalent ions while Na is monovalent. Sodicity can be quantified as exchangeable sodium percentage (ESP); i.e., the exchangeable Na+ expressed as a percentage of cation exchange capacity. If the SAR of the soil equals or exceeds 13, or if the ESP of the soil equals or exceeds 15%, the soil is termed sodic.
(3) ![\begin{equation*}SAR = \frac{[Na^{+}]\;(mmol\;L^{-1})}{(0.5\times[Ca^{2+} + Mg^{2+}])^{\frac{1}{2}}\;(mmol L^{-1})}\end{equation*}](https://openpress.usask.ca/app/uploads/quicklatex/quicklatex.com-1ea99b597ff72e76da6a47e5544acf88_l3.svg#fixme "Rendered by QuickLaTeX.com")
(4) ![\begin{equation*}ESP = \frac{[Na^{+}]\;(cmol(+)\;kg^{-1})}{CEC\;(cmol(+) kg^{-1})} \times 100\end{equation*}](https://openpress.usask.ca/app/uploads/quicklatex/quicklatex.com-4570ddebd5885cda1a971331919ee963_l3.svg#fixme "Rendered by QuickLaTeX.com")
Development of Salinity in Soils
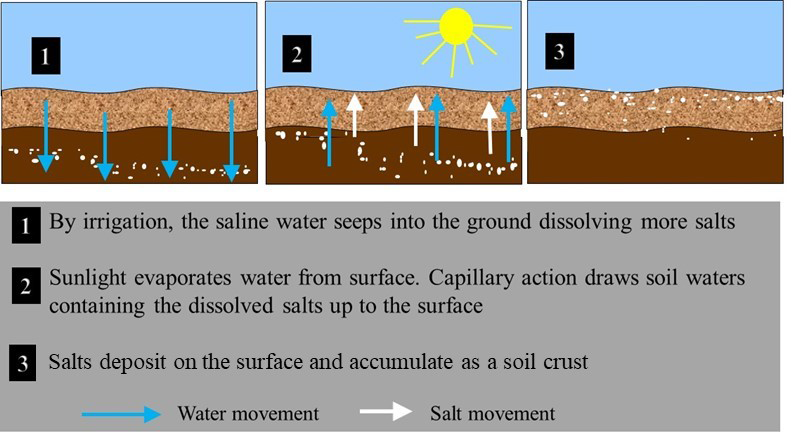
Salts in soils originate from natural and anthropogenic sources. Natural sources include weathering of primary minerals, atmospheric deposition, sea water intrusions, discharge of saline groundwater, and brines from natural salt deposits. Major anthropogenic sources of salts include irrigation with waters containing high salt concentrations (see Figure 5.11), fertilization with synthetic and organic fertilizers with high salt content including manure, biosolids, and compost. Road salts applied during winter in Canada to melt ice and improve road conditions, are also a localized source of salinity to soils.
Soluble salts accumulate in soils for various reasons:
- Dry climatic conditions – Soluble salts accumulate in arid and semiarid areas where rainfall is not sufficient to leach the salts through the soil profile. High evaporation losses from these soils result in the upward movement of water, which may bring salts from the deeper soil layers that then remain at the soil surface as visible crusts when water evaporates. Evaporative losses can range from 50–90% in arid regions resulting in 2- to 20-fold increase in levels of soluble salts.
- Drainage – Poor drainage (due to high water table or low subsoil permeability) can increase the salt content.
- Irrigation water quality – Salinity problems are common in irrigated lands as most irrigation waters contain varying concentrations of soluble salts. The water added to the soil is utilized by the crop or evaporates directly from the moist soil, leaving the dissolved ions behind to form saline crusts.
- Coastal regions – Soils in coastal areas may have high salt accumulation through sea water intrusions and saline groundwater.
- Other management practices – Salt accumulation may take place due to other management practices such as overuse of synthetic fertilizers, continuous application of manures, or yearly application of road salts that are washed into road-side ditches and/or blown onto nearby fields and vegetation by wind.
Measurement of Soil Salinity
The total dissolved salt concentration of a soil can be measured by extracting a soil sample with water and measuring the concentrations of individual salts in the extract. In practice however, measuring individual salts is time consuming, cumbersome and expensive; thus the electrical conductivity (EC) of a solution, which increases in direct proportion to the overall abundance of dissolved salts, is employed as an alternative index to assess and describe soil salinity. Soil extract EC is the preferred measurement to assess the overall salinity status of the soil. The measurement of EC—accomplished with a conductivity meter in soil testing laboratories—is reliable, rapid and cost effective. The EC of a soil is determined in a saturated extract (i.e., in the soil solution extracted from a saturated soil paste) and expressed in units of decisiemens per meter (dS m-1) or millisiemens per centimeter (mS cm-1). Soils with an EC value of saturated soil extract >4 dS m-1 are classified as saline.
Properties of Salt-Affected Soils
Salt-affected soils can be classified as saline, saline-sodic or sodic, depending on the total concentration of Na+ salts in the soil. Saline soils have high salt content but low concentrations of Na+. Sodic soils have relatively low salt content but high concentrations of Na+. Saline-sodic soils have high concentration of both salts and Na+. Figure 5.12 illustrates the classification of salt-affected soils based on the EC, SAR and ESP.
Saline soils have high concentrations of total dissolved salts, with EC values greater than 4 dS m-1 (Figure 5.12). These soils have relatively low amounts of Na+ in the soil solution or on the exchangeable complex, and thus SAR values are less than 13 and ESP values are less than 15%. The soil pH of saline soils is usually less than 8.5. The high salt concentrations in these soils promote flocculation of soil colloids (i.e., individual colloids are attracted together forming flocs or clusters).
Sodic soils have low total concentrations of dissolved salts, with EC values less than 4 dS m-1. These soils, however, have relatively high concentrations of Na+ in the soil solution and on the exchange complex relative to Ca2+ and Mg2+. Therefore, SAR values are greater than 13 and ESP values are greater than 15% (Figure 5.12). Sodic soils typically have pH values greater than 8.5 due to the hydrolysis of Na2CO3. The colloids in these soils are dispersed due to the swelling in response to the high proportion of exchangeable sodium. Swelling and dispersion is greater when the exchange complex has a greater proportion of hydrated, monovalent ions particularly Na+. When hydrated Na+ ions come between clay particles, the electrostatic forces that bind the clay particles together are neutralized and additional layers of water molecules force the particles apart, thus dispersing the soil matrix materials. The high pH also disperses soil organic matter. Alkaline sodic soils classified as Black Solonetz have dispersed, dark coloured humic material coating the soil peds.
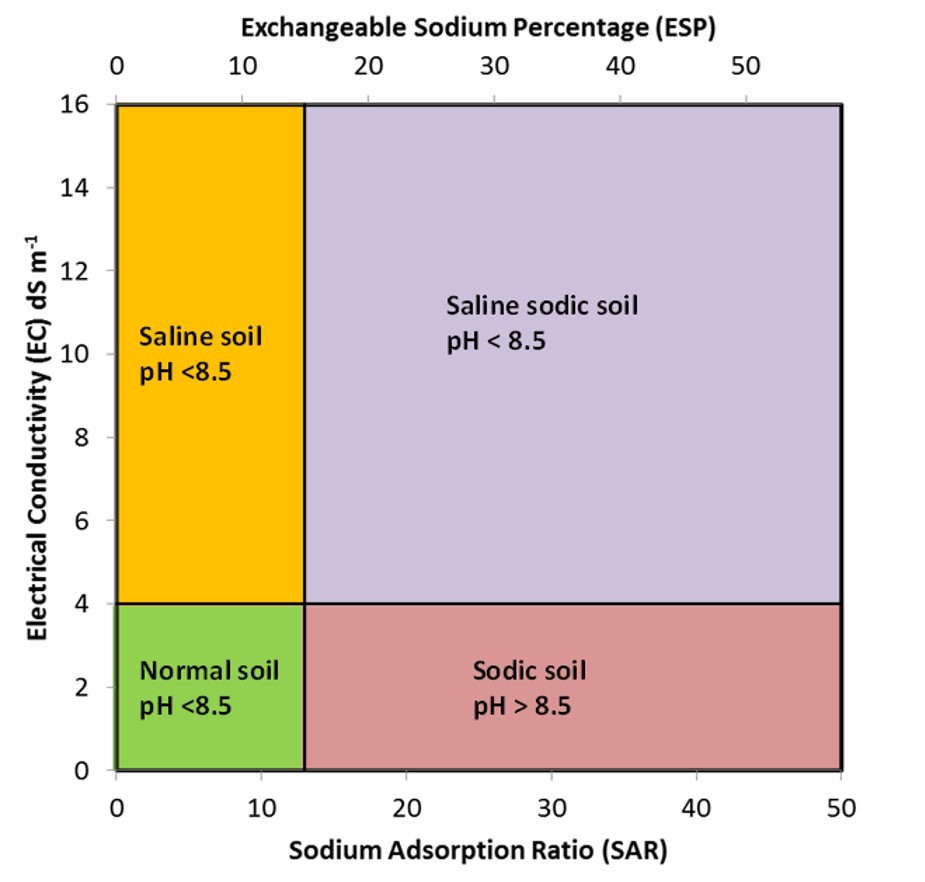
Soils having high concentrations of dissolved salts as well as high concentrations of Na+ are termed saline-sodic soils. Such soils have EC values greater than 4 dS m-1, SAR values greater than 13 and ESP greater than 15%. Saline-sodic soils usually have pH values less than 8.5. These soils are flocculated because of the high concentration of dissolved salts (Sparks, 2003).
Effects of Soil Salinity and Sodicity on Ecosystem Eealth
Soil salinization, one of the most devastating global environmental concerns, is a major cause of land degradation and contributes to desertification of dry ecosystems. Worldwide, salinization of soils leading to salinity and sodicity have caused major reductions in cultivated areas and crop productivity.
Both salinity and sodicity affect soil properties, plant growth, and microbial activity. High salt concentrations will adversely affect plant growth as the dissolved salts lower the osmotic potential of the soil solution. Since plant roots take up water primarily by osmotic mechanisms, they are unable to absorb water from highly saline soils, with some plants being more sensitive to saline conditions than others. This resultant condition is known as “physiological drought” since plant roots are unable to take up water even when the water content in the soil may be adequate for normal growth. Similarly, microbes in the soil may be affected because of an inability to obtain water due to osmotic effect.
Table 5.4. Sensitivity of some plants to saline conditions
| Sensitive | Moderately Sensitive | Moderately Tolerant | Tolerant |
|---|---|---|---|
| (EC < 4 dS m-1) | (EC 4-7 dS m-1) | (EC 7-9 dS m-1) | (EC 9-12 dS m-1) |
| Vegetables | Alfalfa | Fescue | Barley |
| Maple | Corn | Aspen | Wheat |
| Tomatoes | Grasses | Oats | Willow |
| Potatoes | Clover | Soybean | |
| Note: plants capable of surviving EC in the range of 12–15 dS m-1 are considered halophytes; however, very few, if any, plants can survive EC values >15 dS m-1 | |||
The high concentration of sodium in sodic soils favours swelling of expandable clay minerals and dispersion of clay particles, resulting in poor soil structure, reduced permeability and infiltration due to blockage of the soil pores, and formation of surface crusts. Low infiltration and permeability may also increase soil erosion and may lead to saturation of the soil surface during irrigation and rain/snowmelt events, in turn restricting air entry into the soils. This leads to anaerobic conditions in soils which also negatively affects plant growth.
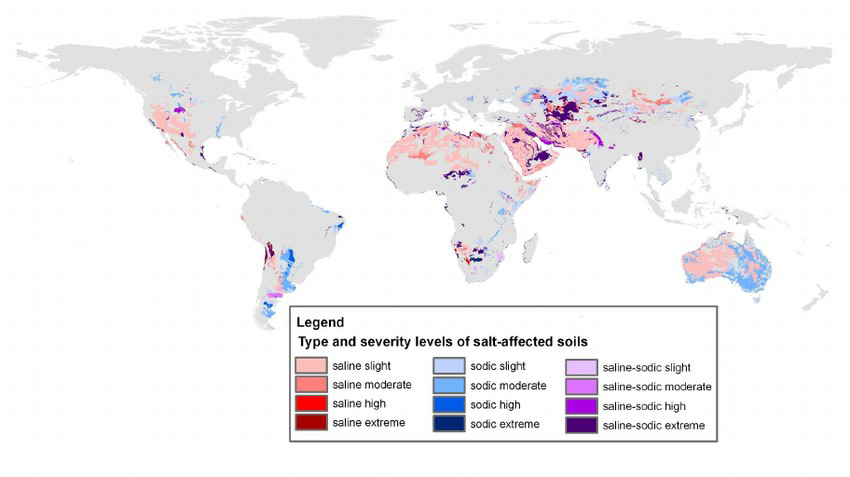
Salt-Affected Soils: Distribution Globally and in Canada
Soil salinization is a global problem. Salt-affected soils occur on all continents, mostly in arid and semi-arid areas. Salinization is of lesser concern in humid areas as rainfall is sufficient to leach excess salts from the soil profile. Salinized areas in the world increase at a rate of about 10% annually because of natural and anthropogenic causes. Based on the 1970–1980 world soil map, the total area of salt affected soils worldwide is estimated as 397 million ha of saline soils and 434 million ha of sodic soils (FAO, 2020). Figure 5.13 illustrates the distribution of saline and sodic soils worldwide (Zheng, 2014). Globally land degradation due to salinization has been reported in the Aral Sea Basin (Amu‐Darya and Syr‐Darya River Basins), the Indo-Gangetic Basin in India, the Indus Basin in Pakistan, the Yellow River Basin in China, the Euphrates Basin in Syria and Iraq, the Murray-Darling Basin in Australia, and the San Joaquin Valley in the United States (Qadir et al., 2014).
There are an estimated 6 to 8 million ha of salt-affected soils in Canada, located primarily in the Canadian prairies (Miller and Brierley, 2011). In 1995, the risk of salinization was rated as moderate to very high affecting an estimated 34% of farmland area in the Prairie Provinces (Acton and Gregorich, 1995), with the estimate decreasing to 12% of farmland area by 2001 (Eilers et al., 2010). This decrease in the risk of salinization potential in agricultural lands in the Prairie Provinces is largely due to the reduction in summer fallow, and conservation practices such as conversion to permanent cover.
Can You Dig It!
Surface and subsurface salinity in the Canadian Prairies – A threat to soil quality and sustainable ecosystem productivity
Soil salinization is a typical process for the Canadian prairies and adjacent areas of the Boreal Plains. It is estimated that at least 1,431,00 ha in the region are affected by salinity. The two images below show white saline crust, which often occurs adjacent to shallow wetlands (sloughs) on the prairies. The salts are dominantly calcium and magnesium sulphates (gypsum and epsom salts) derived from evaporation of saline groundwater discharge from the bedrock below the regional glacial till veneer. The surface expression is the result of a gently rolling topography combined with a high water table in the wetland.


The image to the right shows the effects of subsurface salinity. The most common expression of salt-affected soils is irregular growth of crop plants across a field. The soil surface may look normal, but salinity is often greater at depth in the root zone. Plants typically germinate and grow after seeding due to available moisture at the surface. As the soil dries and plant water consumption increases, the roots penetrate deeper searching for water. At some depth they can encounter higher levels of salt in the subsoil, which acts as a barrier to accessing moisture. The result is stunted or irregular plant growth, and possibly death due to lack of moisture.
Reclamation of Salt Affected Soils
Methods of removing salts from surface soils include scraping, surface flushing and leaching. Since the salts are highly soluble, leaching with good quality water is the most effective and commonly used method of reclaiming saline soils. While saline soils are aggregated and have high infiltration rates, sodic and saline-sodic soils are usually dispersed and have low infiltration rates. Thus, it is difficult to leach the salts and sodium with water from sodic and saline-sodic soils since they are typically dispersed. Thus, to reclaim sodic and saline-sodic soils, application of soil amendments such as gypsum (CaSO4.2H2O) is usually needed. The added Ca2+ promotes soil aggregation and improves water infiltration when monovalent Na+ is replaced by divalent Ca2+. The displaced Na+ and excess salts can then be leached by irrigation with good quality water. However, because leached salts may accumulate in the groundwater, saline drainage water may be diverted through subsurface drains. Other ways of removing salts from salt-affected soils include growing salt tolerant crops (e.g., barley, sunflower) and planting trees that will take up the dissolved cations and anions from soil solution. Preventing salinization by using best management practices (e.g., use of good quality irrigation water in agricultural lands, managing water below root zone through subsurface drainage) is a better approach than allowing salinity to increase in soils and then attempting to reclaim the salt-affected soils.
OXIDATION-REDUCTION (REDOX) REACTIONS
Oxidation-reduction reactions, or redox reactions, involve the transfer of electrons and the conversion of an element from one valence state to another. Understanding the behaviour of redox sensitive elements in soil is central to understanding the behaviour of Gleysolic and Organic soils, and gleyed soil subgroups that occur within all mineral soil orders in Canada (SCWG, 1998). Internationally, about 130 million ha of land, accounting for about 10% or all arable land on the planet, is temporarily flooded for rice production generating reduced soil conditions and emphasizing the importance of understanding redox reactions (IRRI, 1997).
Redox reactions are typically written as half reactions (i.e., the electron and change in oxidation state are explicitly stated in the equation). The reversible reaction is always to be written as a reduction reaction with the electron (e–) on the left side of the equation:
oxidants + e– ↔ reductants
where oxidants are chemical species capable of accepting electrons and reductants are chemical species capable of donating electrons. For example:
Fe3+ + e– ↔ Fe2+
Redox measurements are useful to provide an understanding of chemical behaviour in wet and poorly drained soils (e.g., Gleysolic or Organic soils and gleyed subgroups; see Figure 5.14) as a high redox potential (>300 mV) indicates good availability of O2 (aerobic conditions), whereas a low redox potential (<300 mv) indicates limited availability of O2 (anoxic conditions).

Key redox sensitive elements in soil include oxygen (O2), nitrogen (NO3–, NO2–, NOx, N2O, N2), manganese (Mn4+, Mn2+), iron (Fe3+, Fe2+), sulphur (SO4–, S2-), and carbon (CO2, CH4). These are the main species commonly involved in redox reactions in soils and constitute a reaction sequence indicating the tendency of each element to become reduced:
Oxygen > Nitrogen > Manganese > Iron > Sulphur > Carbon
Typically, elements on the left of this sequence dominate the prevailing oxidation state of the other elements to the right of it. For example, the presence of oxygen dominates electron acceptance over all other elements in the sequence and thus dictates that each of the other species must donate their electrons and be converted to their oxidized form. Thus, in the presence of oxygen (i.e., oxidizing environments) the stable valences of the other elements are NO3–, Mn4+, Fe3+, SO42-, and CO2.
Table 5.5. Half reactions, standard electrode potentials (E°), typical Eh range in soils and mediating micro-organism group for dominant redox species in soil
| Half Reaction | E° (millivolts) | Eh range (millivolts) | Micro-organism |
|---|---|---|---|
| O2(aq) + 4 H+ + 4 e- ↔ 2 H2O | 1230 | 600 to 350 | Aerobes |
| 2 NO3- + 12 H+ + 10 e- ↔ N2(g) + 6 H2O | 1240 | 400 to 200 | Denitrifiers |
| Mn(VI)O2(s) + 4 H+ + 10 e- ↔ Mn2+ + 2 H2O | 1220 | 300 to -50 | Mn-reducers |
| Fe(III)(OH)3(s) + 3 H+ + e- ↔ Fe2+ + 3 H2O | 930 | 50 to -50 | Fe-reducers |
| SO4 + 9 H+ + 8 e- ↔ S2- + 4 H2O | 250 | -150 to -200 | Sulphate reducers |
| CO2(aq) + 8 H+ + 8 e- ↔ CH4(aq) + 2 H2O | 170 | -150 to -250 | Methane fermenters |
The notation used to identify redox species in in the aqueous phase (i.e., soil solution) is different than that use to identify elements occurring in the solid phase. For example, iron species in the aqueous phase are typically noted as Fe2+ and Fe3+, whereas iron species present in soil minerals and other solids are expressed as Fe(II) and Fe(III).
Generally, oxidative decomposition (decay) of organic matter is mediated by various microorganisms to derive energy (see Table 5.5). Consequently, organic matter is the primary source of electrons in soil. Conversion/decay of organic matter varies considerably from complete conversion to CO2 (gas) to partial and incomplete oxidation and conversion to intermediate products (i.e., conversion to humus), but all involve the reduction of redox species to their reduced form. Because the composition of soil organic matter is highly complex (see Chapter 3) there is no general chemical formula attributed to it (van der Park, 2006).
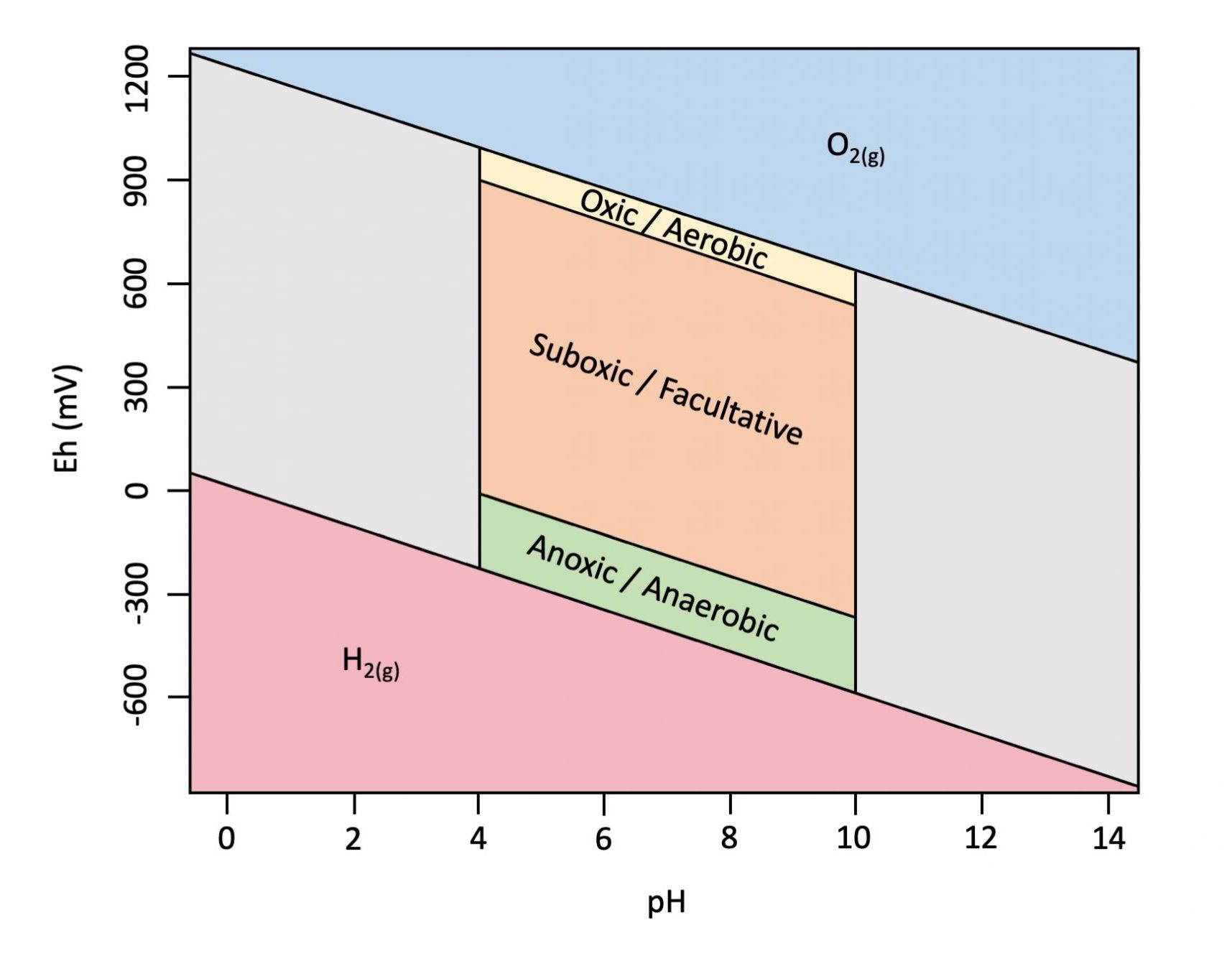
Figure 5.15 is an example of an Eh-pH diagram, or Pourbaix diagram, a graphical representation showing the approximate ranges for Eh and pH in soil. Soil pH values generally range from about 4 to 10, with Eh bounded on the top and bottom by the redox stability of water. The Eh varies with pH according to Nernst equation by -59.16 mV per pH unit at 25 ℃ (298.15K). Figure 5.15 also indicates the approximate ranges for oxic conditions (O2 reduction reaction is dominant and mediated by aerobic organisms); sub-oxic conditions (nitrogen, iron and manganese reduction reactions are dominant and mediated primarily by facultative organisms); and anoxic conditions (sulphur and carbon reduction reactions are dominant and mediated by anaerobic organisms) in soil.
Oxygen
In non-saturated (aerobic) soils, oxygen occurs in both the soil gas phase (O2 g) and in the soil solution (aqueous) phase (O2 aq). Although similar in composition to that of the atmosphere (about 20% O2 by volume), the concentration of oxygen in soil air is typically somewhat lower as oxygen in the soil gas phase is constantly consumed by plant roots and microbial respiration. Under oxygenated (non-saturated) conditions, oxygen is effectively in infinite supply capable of accepting electrons and completely oxidizing soil organic carbon to CO2 (g). Continued introduction of oxygen to topsoil due to cultivation practices is the primary reason for the reduction in soil organic matter levels as a result of oxidation reactions.
Although the concentration of oxygen in the soil gas phase is usually high, the concentration of dissolved oxygen in water is far less, being generally on the order of 10 mg L-1 (at 15 ℃) but varying with temperature. As dissolved oxygen becomes consumed, it is replenished by the normally abundant pool of oxygen in the soil gas phase. Aside from the large difference in the abundance of oxygen, the mobility of oxygen is roughly 10,000 times slower in water compared to oxygen in the gas phase. Consequently, as soils become increasingly saturated with water the oxygen supply from air becomes increasingly limiting until all, or almost all, of the soil pore space becomes filled with water, and oxygen transport is possible only via the aqueous phase. The redox potential decreases as the soil effectively becomes sub-oxic or even anoxic/anaerobic (see Figure 5.15).
Can You Dig It!
Seasonally anoxic soils during spring snowmelt flooding

Most farmlands in the Canadian prairies get flooded during the spring snowmelt period. Seasonal flooding can make the soil sub-oxic or anoxic as the redox potential decreases. This will have many environmental impacts. For example, phosphorus concentration in floodwater may increase when Fe(III) and Mn(IV) are reduced to divalent forms releasing the phosphate associated with them. Released phosphorus may end up in lakes causing algal blooms. Anoxic conditions may also enhance emissions of greenhouse gases such as nitrous oxide (N2O) and methane (CH4).
Nitrogen
The primary reason for the depletion of available oxygen from soil is saturation with water. As discussed above the amount of dissolved oxygen in soil water is very limiting and diffusion of oxygen through water is very slow. As oxygen becomes limiting/depleted, typically under temporary saturated conditions, nitrate-reducing microbes become active initiating the process of denitrification. The process of denitrification is a major pathway for the loss of fertilizer nitrogen applied as a nutrient source for crops. Nitrate (NO3–) is reduced to nitrogen gas (N2) with nitrite (NO2–), nitric oxide (NO) and nitrous oxide (N2O) as intermediate species. Soluble nitrite is a short-lived aqueous intermediate while nitric oxide (NO) and nitrous oxide (N2O) intermediates are involved in the destruction of stratospheric ozone (O3) layer. Nitrous oxide is also an important greenhouse gas with a global warming potential (GWP) 265–298 times that of carbon dioxide.
Manganese
As nitrate becomes depleted, manganese reducing bacteria take over. Although the total content of manganese in soil is typically only about 750 to 1000 mg kg-1 (Ure and Berrow, 1982; Bowen, 1979) it is still a major redox species in soil. Manganese exists in soil mainly as Mn2+, or as Mn(VI). The soluble form prevalent in anoxic soil is Mn2+ (aq), which when oxygen is introduced oxidizes and precipitates manganese as Mn(IV), usually as non-crystalline (amorphous) oxy-hydroxides. Manganese oxides appear as black coatings on soil particles, or as black nodules, some as large as 2 cm in diameter. When present, manganese solids act as scavengers for nutrient anions (e.g., phosphate).
Iron
Iron is the fourth most abundant element in soil and is responsible for diverse mottles characterized by the drastic differences in colouration between reduced (Fe2+) and oxidized (Fe3+) valences in wet and water-logged soils. Strongly reduced soils with Eh values in the range of -200 mV typically have gleyed (dull grey or practically white) colour due to the presence of reduced iron. This reduced soil condition means that dissolved oxygen as well as nitrate and manganese are essentially absent and that most dissolved iron is present as the Fe2+ species, with most iron compounds (responsible for the grey colour) occurring as Fe(II) oxides and hydroxides (e.g., ferrous hydroxide [Fe(OH)2]. When the water table lowers or oxygenated water infiltrates into a reduced soil (perhaps through a root channel, open pore or fracture), ferrous iron transfers electrons to the oxygen resulting in the formation of oxidized [Fe(III)] compounds. These are observed as mottles occurring as reddish blotches on an otherwise dull grey background. Mottles are a very common morphological feature in Gleyed sub-groups or Gleysolic soils that are periodically wet or have a fluctuating water table. Mottles are diagnostic features in the identification of moderately well, imperfectly, and poorly drained soils.
Sulphur
Although there are several oxidation states for sulphur, only two oxidation states are prevalent in soil. Under aerobic conditions sulphate (SO42-) is the stable form with an oxidation state of +6, while sulphide (S2-) with an oxidation state of -2 is the stable form under strongly reducing conditions (McBride, 1994). Sulphate is the primary form of sulphur taken up by plants, precipitating primarily as the mineral gypsum (CaSO4•2H2O) when there is sufficient dissolved calcium (Ca2+) present in the soil solution. Sulphate may be reduced to sulphide which has very low solubility in water under anoxic conditions forming either odourous hydrogen sulphide gas (H2S with the aroma of rotten eggs), or insoluble metal sulphides such as FeS2 if soluble metals are present.
The introduction of oxygen into sulphide containing soils results in the re-oxidation of metal sulphides:
FeS(s) + 15/4 O2 + 7/2 H2O ↔ Fe(OH)2 + 4H+ + 2 SO42-
This reaction is common in reclaimed coastal soils high in sulphides, in Northern Prairie soils derived from sulphide containing shales (referred to as acid sulfate soils) and in mine wastes (tailings and waste rock) containing pyrite and other metal sulphides.
Carbon
Ultimately if sulphate is limited, then methane producing bacteria continue the process of organic matter decomposition under anaerobic conditions as follows:
2CH2O ↔ CO2 + CH4
Methane production typically occurs under stagnant, water saturated conditions in wetlands. Under these conditions the organic decay process is slow and incomplete, resulting in the accumulation of soil organic matter and the formation of peatlands and organic soils.
Measurement of Redox
The direct measurement of redox (oxidation-reduction) potential in soil is a quandary. Fundamentally, redox measurement is extremely simple, being similar to pH measurement in mechanics, but very different from pH with respect to fundamental chemistry and interpretation. Redox measurements or measuring the “concentration of electrons” is not analogous to pH because free electrons do not exist in soil solutions in the same way as hydrogen (H+) ions. Redox is typically measured as a voltage (usually reported as volts or millivolts), which represents a measure of the potential for electrons to be transferred from one element to another. This potential measure provides an indication of which redox species are the most receptive to gaining electrons at the time of the measurement. Actual times for electron transfer are typically much slower than the time required to complete a redox measurement, with reactions being mediated by specific biological organisms as discussed above. The redox measurement thus indicates the tendency or likelihood that a redox reaction will occur, while the reaction itself may not truly occur to any meaningful extent as the measured redox condition may be only temporary.
Regardless of difficulties with measurement, redox measurements provide critical data for the description and analysis of wet or seasonally saturated soils. Measurements are actually recordings of voltage (Eh) between a reference electrode and a sensor electrode inserted into a soil. The most common sensor electrodes are usually constructed of platinum wire (Pt electrode). The voltage results from an exchange of electrons between a redox couple such as ferrous (Fe2+) and ferric (Fe3+) iron during the process of reduction and oxidation. In soils that have fluctuating wet and dry conditions, wide seasonal fluctuations in Eh values occur (Fiedler et al., 2007).
Although free electrons do not exist in soil, the concept of free electrons—analogous to describing free-proton activity and pH values—is useful as a first step in describing and understanding soil redox status. Similar to pH, “electron activity” may be express as pe (analogous to pH) which is the negative common logarithm of electron activity:
(5) 
Values for pe are not measured directly, but derived from Eh through the Nernst equation:
(6) 
where Eo is the standard electron potential for the half-reaction, R is the molar gas constant (8.31 joules/mole/K), T is absolute temperature (298.15 K); F is the Faraday constant (96,490 coulombs/mole) and n is the number of electrons transferred in the half reaction.
Although the measurement of Eh using a platinum electrode is a simple procedure, it is also subject to measurement uncertainty. Principle issues (Sposito, 1989) include:
- voltage measurement in soil is usually due to more than one-half reaction;
- concentrations of redox species in solution may be too small to provide electron transfer detectable by the Pt electrode;
- several redox species are not readily reactive;
- Pt electrodes are easily contaminated or ‘poisoned’ by oxides or other coatings; and,
- the salt content of the soil solution (and its ability to conduct electrons) may differ greatly from the calibration buffer.
These uncertainties mean that Eh measurements are considered qualitative measures that provide a general indication of the soil redox status sufficient to differential oxic, suboxic, and anoxic environments (Figure 5.14). The Eh data should not be used to numerically quantify the concentrations of redox species but can be used to determine which redox couples are most active at the time of measurement. Quantitative measurements for redox species should involve direct measurement of the species of interest; for example, dissolved oxygen or ion specific electrode measurement, or multi-species techniques such as ion chromatography.
SUMMARY
- Soil chemistry involves the study of the chemical composition, chemical properties and chemical reactions involving soil solids (organic matter and inorganic minerals), liquids (soil water), gases (soil air), including interactions with plants and soil organisms. Soil chemical properties have huge implications for global human and environmental health. Through the influence on fate of nutrients and contaminants, soil chemical properties play a role in crop production and food security, biodiversity, water and air pollution, global climate change through greenhouse gas emissions etc. Knowledge of soil chemistry is fundamental in understanding the fate of nutrients and contaminants in the soil system, predicting their environmental impact and finding solutions to mitigate impacts.
- Soil chemical properties are largely influenced by the type and quantity of inorganic minerals present in the soil and the organic matter (humus) content. Inorganic colloids mainly include phyllosilicates (layer silicate minerals), amorphous aluminosilicates and oxyhydroxide minerals.
- The presence of surface charges in both inorganic minerals and humus particles is responsible for ion adsorption and exchange in soils. Most charges are permanent (not dependent on soil pH) and negative, while others vary with pH and may be negative or positive.
- Negative charges on soil particles can attract and adsorb cations, which can be released back to the soil solution through ion exchange. Cation exchange capacity, defined as the sum of total exchangeable cations that a soil can adsorb, is an important soil chemical property that influences the retention of cationic plant nutrients supporting plant growth and retention of cationic pollutants minimizing groundwater pollution.
- Soil pH is a measure of acidity and alkalinity, which controls many of the chemical processes that take place in the soil including availability of plant nutrients. The primary factor influencing the natural soil pH are the geological parent material from which soils are derived. Liming is a practice commonly used to raise the pH of acidic soils.
- Soil salinity gives an indication of the salt content of the soil and is determined by measuring the electrical conductivity of a saturated soil paste extract. Depending on the electrical conductivity and the relative concentration of sodium in soil solution or exchangeable complex, salt-affected soils are classified as saline, saline-sodic and sodic soils. Irrigation with saline waters, particularly in arid climates, is one of the major anthropogenic causes of salinity. Excess salinity in soil can decrease plant available water and affect plant growth.
- Oxidation-reduction (redox) reactions in soil involve the conversion of an element from one valence state to another. Key redox sensitive elements in soil include oxygen (O2), nitrogen (NO3–, NO2–, NOx, N2O, N2), manganese (Mn4+, Mn2+), iron (Fe3+, Fe2+), sulphur (SO42-, S2-), and carbon (CO2, CH4). Redox reactions influence dynamics for certain plant nutrients (eg., nitrogen, sulfur, iron, manganese) and gaseous losses from soils, including greenhouse gas emissions (nitrous oxide and methane).
STUDY QUESTIONS
- Explain why montmorillonite has a higher shrink-swell capacity than kaolinite.
- Define isomorphous substitution. Briefly explain how isomorphous substitution causes negative charge development on clay mineral surfaces.
- Explain why Vertisols in Canada are typically very sticky when wet and form wide cracks when dry.
- Define cation exchange capacity. Why is soil cation exchange capacity important in protecting and enhancing biodiversity and ecosystem health?
- Consider the structure and properties of phyllosilicates summarized in Figure 5.5 and answer the following True/False questions.
- Presence of interlayer hydrogen bonds makes kaolinite a relatively non-expandable clay mineral.True or False
- 2:1 type clay minerals are composed of one tetrahedral sheet sandwiched between two octahedral sheets.True or False
- There are no interlayer hydrogen bonds in 2:1 type clay minerals since hydroxyl groups are not exposed.True or False
- Montmorillonite and illite (hydrous mica) are both 2:1 type phyllosilicates. Montmorillonite freely expands while illite has a limited expandability.True or False
- Illite (hydrous mica) has a high layer charge due to isomorphous substitution but its cation exchange capacity is relatively low.True or False
- A soil with a cation exchange capacity of 42 cmol(+) kg-1 has the following cations on the exchange complex: Na+ = 7 cmol(+) kg-1 soil, Ca2+= 12 cmol(+) kg-1 soil, K+= 6 cmol(+) kg-1 soil, and Mg2+= 8 cmol (+) kg-1 soil). Calculate the base saturation of this soil.
- Explain how a change in soil pH influences the cation and anion exchange capacities of soils.
- Differentiate between active and reserve acidity.
- “Soil pH is greatly influenced by the parent material from which soil is formed”. Explain the statement giving examples of parent materials in Canada to support your answer.
- Briefly explain how soil colloids buffer soil pH changes caused by acidic or basic inputs, such as nitrogen containing fertilizers and liming amendments.
- Briefly explain how soil pH influences nutrient availability to plants.
- Define saline, sodic and saline-sodic soils. Outline their major characteristics and effects on ecosystem health.
- Briefly explain how irrigation under arid climates can influence soil salinity development.
- A soil with a cation exchange capacity of 20 cmol(+) kg-1 has exchangeable Na content of 8 cmol(+) kg-1.
- Calculate the exchangeable sodium percentage (ESP) of the soil.
- If the electrical conductivity of the saturated extract of this soil is 5.6 dS m-1 (decisiemens per meter), how would you classify the soil? (Hint: See Figure 5.11)
- How can such a soil be reclaimed?
- Identify the key redox sensitive elements in the soil. Give one oxidized form and one reduced form for each of these elements.
- Describe how the redox state of the soil influences nutrient availability and greenhouse gas (nitrous oxide and methane) emissions.
WORKED EXAMPLES
Describe the different mechanisms by which negative charges are developed on surfaces of soil colloids.
Two main mechanisms are responsible for the development of negative charges to surfaces of soil colloids. One is isomorphous substitution in phyllosilicate minerals where central cation in the octahedral or tetrahedral sheet in the mineral is substituted by other cations. The substitution occurs with ions of similar sizes and a negative charge is developed when a higher valent cation is replaced by a lower valent cation. For example, if the Si4+ in the tetrahedral sheet is replaced by Al3+ (as in illite or hydrous mica), it will give rise to a surface negative charge. Similarly, in the octahedral sheet, Al3+ may be substituted by Mg2+ which creates a negative charge on the mineral. The negative charges due to isomorphous substitution is permanent and not dependent on pH.
The second mechanism of development of negative charges on soil colloids is the deprotonation (dissociation) reactions depending on pH. Oxyhydroxide clays and the edges of phyllosilicate and amorphous silicate clays have hydroxyl groups which can dissociate at high pH developing a negative charge on surface. Organic colloids (humus) also can carry pH dependent negative charges due to dissociation of functional groups such as carboxyl and phenolic groups. These charges increase with increasing pH.
Explain why the clay mineral montmorillonite has a higher CEC than kaolinite (hint: see Figure 5.2)
Montmorillonite is a 2:1 phyllosilicate clay mineral with 2 silicon tetrahedral sheets enclosing an aluminum octahedral sheet. Isomorphic substitution, e.g. of magnesium (Mg2+) for some of the aluminum (Al3+) in the octahedral sheet results in a permanent negative charge. In addition, montmorillonite clays expand when wetted, as water enters the interlayer space between adjacent tetrahedral sheets. This increases the available surface area for cation adsorption, since in montmorillonite has both external & internal surfaces available for cation adsorption. The combination of net negative charge due to isomorphic substitution and high specific surface area gives montmorillonite a high CEC.
Kaolinite, on the other hand, is a 1:1 phyllosilicate clay mineral, which consists of single silicon tetrahedral sheets alternating with single aluminum octahedral sheets. There is very limited isomorphic substitution, and therefore the kaolinite has a limited number of negative charges. Additionally, being a 1:1 phyllosilicate clay mineral, which does not expand, kaolinite has lower effective surface area on which cations can be adsorbed. Thus kaolinite has a lower CEC.
Fill in the characteristics of the phyllosilicates in the table below.
| Mineral | Type | Isomorphous substitution | CEC | Surface area | Expandability |
|---|---|---|---|---|---|
| (1:1, 2:1, 2:1:1) | (Low/ High) | (Low/ High) | (Low/ High) | (Low/ High) | |
| Kaolinite | 1:1 | Low | Low | Low | Low |
| Montmorillonitee | 2:1 | High | High | High | High |
| Illite (hydours mica) | 2:1 | High | Low | Low | Low |
| Chlorite | 2:1:1 | High | Low | Low | Low |
Calculate the CEC of the soil
A soil has 3% organic matter, 20% montmorillonite, and 17% kaolinite with the remaining 60% being sand and silt sized primary mineral grains. Calculate the cation exchange capacity of the soil in cmol (+) kg-1 (Note: assume cation exchange capacity of organic matter = 200 cmol(+) kg-1, montmorillonite = 100 cmol(+) kg-1 and kaolinitie = 10 cmol(+) kg-1).
CEC of the soil = [0.03 x 200 cmol(+) kg-1 ] + [0.20 x 100 cmol(+) kg-1] + [0.17 x 10 cmol (+) kg-1] = 27.7 cmol(+) kg-1
Identify four major factors that contribute to soil acidity and briefly explain their contribution.
Major factors contributing to soil acidity are acidic parent material, high rainfall, application of ammonium fertilizers, and decay of organic matter. Soils derived from granite-based parent materials, commonly referred to as acidic parent materials are generally acidic in reaction. The natural acidity of rainfall, because of the dissolved carbon dioxide, is a cause of soil acidity. In areas of high rainfall basic cations in soils are leached, increasing the acidity. Application of ammonium fertilizers may also contribute to acidity because the nitrification of ammonium release protons. The decay of organic matter release organic acids contributing to soil acidity.
Name two natural and two anthropogenic causes of soil salinity and briefly describe how they contribute to salinity development.
Two natural causes of soil salinity are the dry climatic conditions and poor drainage. Soluble salts accumulate under dry climatic conditions where rainfall is not sufficient to leach the salts through the soil profile. High evaporation losses from the soils may bring the salts from deeper soil layers with the upward movement of water, which remain at the soil surface.
Under poor drainage conditions (due to high water table or low subsoil permeability), salts are not removed through drainage and leaching, which will result in accumulation of salts on the surface when water evaporates from the soil.
Two anthropogenic causes of salinity are irrigation with saline waters, particularly in dry regions, and overuse of fertilizers. Irrigation water will move downward and dissolve more salts. When the evaporation is high under a dry climate the water moving up will bring dissolved salts. Water will evaporate leaving the salts on the surface. Fertilizers (which are salts containing different nutrients) can increase the salt content of soils if used inappropriately.
Consider the following data for four mineral soils that all have a clay loam texture:
| Soil Property | Soil #1 | Soil #2 | Soil #3 | Soil #4 |
|---|---|---|---|---|
| pH | 8.8 | 8.1 | 7.5 | 7.6 |
| electrical conductivity (dS/m) | 2.7 | 12.2 | 1 | 4.3 |
| base saturation (%) | 90 | 90 | 60 | 86 |
| cation exchange capacity | 12 | 11 | 19 | 9 |
| (cmolc/kg) | ||||
| exchangeable sodium (%) or ESP | 17.3 | 16.8 | 2.1 | 2.5 |
| Salinity classification | Sodic | Saline-sodic | Non saline | Saline |
A) Determine the salinity classification (i.e., saline, sodic, saline-sodic, or non saline) for each of these 4 soils. [Hint: see Figure 5.12]
B) Which of the above soil(s) would most likely be in a dispersed state? Briefly explain your answer based on the information provided.
Soil #1 is most likely be in dispersed state since it has a high ESP of 17.3% while the salt concentration is low (with EC of 2.7 dS m-1).
C) What are the implications of a dispersed soil for plant growth?
Since individual soil particles are in dispersed state, there is no flocculation and consequently no aggregate formation. The consequence of this is that soil will have poorly developed soil aggregate structural units and be dominated by micropores, which prevent air & water movement and also impede root growth. Thus, plant growth will be impaired.
Identify the common oxidized and reduced forms of the following elements in soils: manganese, iron, sulfur, and nitrogen.
- manganese: Mn4+, Mn2+ (reduced)
- iron: Fe3+, Fe2+
- sulphur: SO42-, S2-
- nitrogen: NO3–, NO2–, NOx, N2O, N2
REFERENCES
Acton D.F., and Gregorich L.J. 1995. The health of our soils: toward sustainable agriculture in Canada. Agriculture Agri-Food Canada, CDR Unit, Ottawa.
Bowen H.J.M. 1979. Environmental chemistry of the elements. Academic Press, London, New York.
Brady N.C. and Weil, R.R. 2010. The Nature and Properties of Soils 13th Ed. Pearson Education.
Canada Department of Agriculture; Research Branch 1976. Glossary of Terms in Soil Science, Publication 1459.
Eilers W., MacKay R., Graham L. and Lefebvre A. (eds.). 2010. Environmental Sustainability of Canadian Agriculture: Agri-Environmental Indicator Report Series — Report #3. Agriculture and Agri-Food Canada, Ottawa, Ontario.
FAO 2020. Food and Agriculture Organization of the United Nations. FAO Soil Portal. http://www.fao.org/soils-portal/en/
Fiedler S., Vepraskas M.J., and Richardson J.L. 2007. Soil Redox Potential: Importance, Field Measurements and Observations. Advances in Agronomy 94:1-54.
Havlin J.L., Tisdale S.L, Nelson W.L., and Beaton J.D. 2013. Soil Fertility and Fertilizers: An Introduction to Nutrient Management, 8th ed. Upper Saddle River, NJ: Prentice Hall.
Hendershot W.H., Lalande H., and Duquette M. 2008. Soil Reaction and Exchangeable Acidity. Pages 173-178 IN Carter, M.R. and Gregorich, E.G. (eds.) Soil Sampling and Methods of Analysis. 2nd Edition. Canadian Society of Soil Science. CRC Press, Baton Rouge.
Hendershot W.H., Lalande H., and Duquette M. 1993. Soil Reaction and Exchangeable Acidity. Pp. 167-176 in Carter M.R. (ed.) Soil Sampling and Methods of Analysis. 1st Edition. Canadian Society of Soil Science. CRC Press, Baton Rouge.
International Rice Research Institute (IRRI). 1997. Rice Almanac, 2nd Ed. International Rice Research Institute, Manila.
McKenzie R.H. and Woods S.A. 2010. Management of sodic soils in Alberta, Agdex 518-20, Alberta Agriculture. https://www1.agric.gov.ab.ca/$depart...18-20.pdf?Open Element
McBride M.B. 1994. Environmental Chemistry of Soils. Oxford University Press. New York. 406pp.
Miller J.J. and Brierley J.A. 2011. Solonetzic soils of Canada: Genesis, distribution and classification. Can. J. Soil Sci. 91(5):889-902.
Munroe J. (ed.) 2018. Soil Fertility Handbook. Publication 611 3rd edition, Ontario Ministry of Agriculture, Food and Rural Affairs. 236pp.
Newman A.C. and Hayes M.H. 1990. Some properties of clays and of other soil colloids and their influences on soils. In: De Boodt M.F, M.H. Hayes, A. Herbillon (Eds). Soil colloids and their associations in aggregates. Springer Science & Business Media; Boston. MA, USA. pp. 39-55.
Qadir M., Quillérou E., Nangia V., Murtaza G., Singh M., Thomas R.J., Drechsel P., and Noble A.D. 2014. Economics of salt‐induced land degradation and restoration. In: Natural Resources Forum 38 (4):282-295.
Shreeja D. (n.d.) Phosphate Fixation in Soil: 3. Reactions of Anion Fixation. Available at https://www.soilmanagementindia.com/...-fixation/2252
Sparks D.L. 2003. Environmental Soil Chemistry. Academic Press, London, UK.
Sparks D.L. 2019. Fundamentals of Soil Chemistry. Encyclopedia of Water: Science, Technology, and Society, pp 1-11.
Sposito G. 1989. The Chemistry of Soils. Oxford University Press. New York. 277pp.
Soil Classification Working Group (SCWG), 1998. The Canadian System of Soil Classification. 3rd Edition, Research Branch Agriculture and Agri-Food Canada, Publication 1646.
Soil Survey Staff. 2014. Kellogg Soil Survey Laboratory Methods Manual. Soil Survey Investigations Report No. 42, Version 5.0. R. Burt and Soil Survey Staff (ed.). U.S. Department of Agriculture, Natural Resources Conservation Service. 1001pp.
Ure A.M., and Berrow M.L. 1982. The Elemental Constituents in Soils. Pages 94-204 IN Bowen, H.J.M. (ed.). Environmental Chemistry. Volume 2. Royal Soc. Chem. London.
Van der Perk M. 2006. Soil and Water Contamination from Molecular to Catchment Scale. Taylor and Francis, London 389pp.
Zheng T. 2014. Numerical Analysis of Modeling Concepts for Salt Precipitation and Porosity-Permeability Evolution during Brine Evaporation. Doctoral dissertation, Universität Stuttgart, Germany.
About the Authors
Darshani Kumaragamage, Professor, Department of Environmental Studies and Sciences, University of Winnipeg, Manitoba, Canada.

I obtained a BSc and a MPhil in Agriculture from the University of Peradeniya, Sri Lanka, and a PhD in Soil Science from the University of Manitoba, Canada, specializing in soil physical chemistry. I have over 30 years of teaching experience at the University level and have taught a variety of courses in Soil Science including Introduction to Soil Science, Soil Chemistry, and Soil Fertility as well as courses in Environmental Science. My research interests include assessing, monitoring, and mitigating environmental impacts of fertilizers and animal manure, with special focus on phosphorus transformations and mobility in soils under different climatic and management scenarios. I have authored/coauthored 65 peer-reviewed journal publications.
My favorite memory of working with soil was when I conducted a field research in Manitoba on a very sticky, chernozemic, clay soil on one rainy day. I had to take soil samples from many plots, and as I stepped into the field, about one kilo of soil got stuck onto my boots. With the next step, it almost doubled, and with each step I took, my boots got heavier and heavier until I literally had to drag my feet to go from one plot to another.
C. James (Jim) Warren, Ph.D. P.Geo., Land Resource Specialist, Ontario Ministry of Agriculture Food and Rural Affairs, Guelph, Ontario, Canada.

I am a P.Geo. and an environmental soil scientist with a diverse background in research, consulting and teaching. I am a graduate of both the University of Guelph (B.Sc. (Agr), Soil Science), and the University of Alberta (M.Sc. Soil Science; Ph.D. Soil Chemistry). I conducted research in agronomy, geochemistry, and mineralogy at the University of Guelph and University of Waterloo before becoming a private consultant in acid mine drainage. I am currently employed as a Land Resource Specialist conducting soil survey and pedology work with the Ontario Ministry of Agriculture Food and Rural Affairs, and am also an Adjunct Professor with the University of Guelph. I have delivered courses in soil science, soil mineralogy, soil chemistry, geochemistry, and pedology as a sessional lecturer at four universities (Alberta, Guelph, Waterloo, and Toronto). I have authored or co-authored over 100 publications in refereed journals; book chapters, conference proceedings and reports.
How did I get interested in soils? Well, I grew up on a farm where, as a teenager, I spent a lot of time on a tractor doing tillage operations and had a lot of time to think. One of the questions I asked myself during long days in the field was: “What makes plants grow?” With that I spent the next several decades trying to understand soils to answer that question. The quest continutes . . .
Graeme Spiers, Chair of Environmental Monitoring and Professor, School of the Environment, Department of Earth Sciences / Department of Biology, Laurentian University, Sudbury, Ontario, Canada.

I teach in the School of the Environment, the Department of Earth Sciences and the Department of Biology, Laurentian University. I obtained a B.Sc. in Earth Science and Botany at the University of Waikato in New Zealand, and M.Sc. and Ph.D. degrees from the University of Alberta, specializing in Pedology, Clay Mineralogy and Chemistry. With commercial laboratory and consulting experience across Canada, I have been associated with research programs at the Universities of Alberta, Guelph, Waterloo, Laurentian and New Brunswick. My research has resulted in numerous technical and scientific publications and presentations in analytical chemistry, aquatic chemistry, environmental geology, ecology, plant biology and soil science. Prior to attending university on a full-time basis, I was a New Zealand dairy farmer for 10 years, earning my undergraduate degree as a long-term hobby between milkings. In Sudbury, I am involved in research initiatives examining the effects of both historical and current anthropogenic metal emissions on soils, rivers and lakes, as well as vegetation within the Sudbury Smelter Footprint. I am actively involved in a variety of research programs across Canada, and actively collaborates with researchers in Russia (Moscow State University and the Russian Academy of Sciences), Australia, New Zealand, China, Peru and South Africa.